Lin Guan Ning, Cai Zhipeng, Lin Guohui, Chakraborty Sounak, Xu Dong
Digital Biology Laboratory, Informatics Institute, Computer Science Department and Christopher S, Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
BMC Bioinformatics. 2009 Jan 30;10 Suppl 1(Suppl 1):S5. doi: 10.1186/1471-2105-10-S1-S5.
With the increasing availability of whole genome sequences, it is becoming more and more important to use complete genome sequences for inferring species phylogenies. We developed a new tool ComPhy, 'Composite Distance Phylogeny', based on a composite distance matrix calculated from the comparison of complete gene sets between genome pairs to produce a prokaryotic phylogeny.
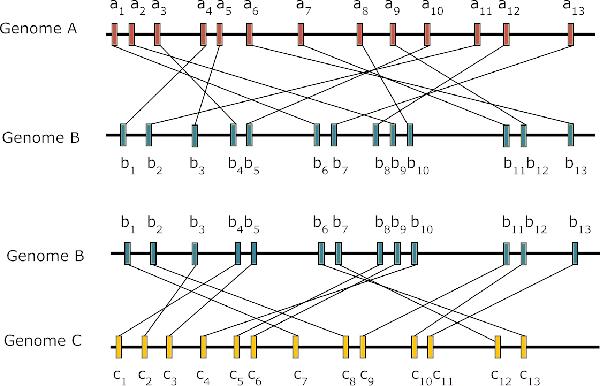
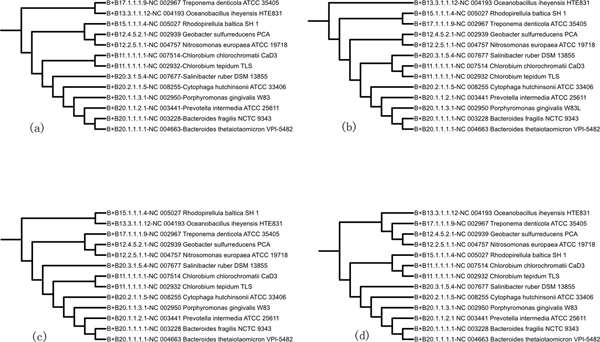
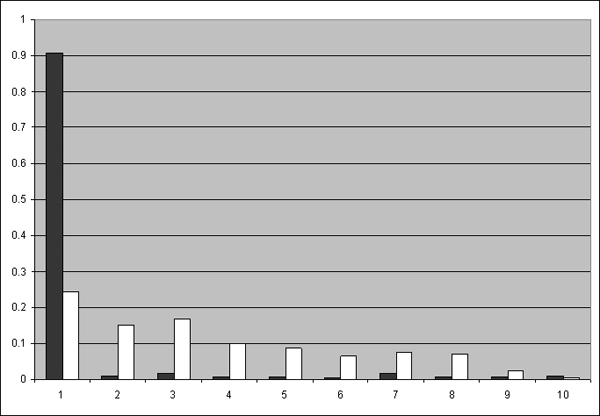
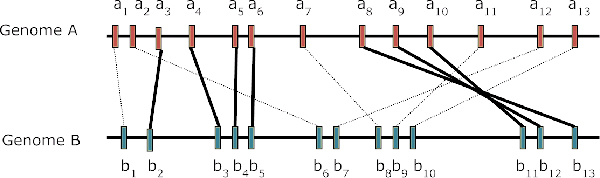
The composite distance between two genomes is defined by three components: Gene Dispersion Distance (GDD), Genome Breakpoint Distance (GBD) and Gene Content Distance (GCD). GDD quantifies the dispersion of orthologous genes along the genomic coordinates from one genome to another; GBD measures the shared breakpoints between two genomes; GCD measures the level of shared orthologs between two genomes. The phylogenetic tree is constructed from the composite distance matrix using a neighbor joining method. We tested our method on 9 datasets from 398 completely sequenced prokaryotic genomes. We have achieved above 90% agreement in quartet topologies between the tree created by our method and the tree from the Bergey's taxonomy. In comparison to several other phylogenetic analysis methods, our method showed consistently better performance.
ComPhy is a fast and robust tool for genome-wide inference of evolutionary relationship among genomes. It can be downloaded from http://digbio.missouri.edu/ComPhy.
随着全基因组序列的可得性不断增加,使用完整基因组序列来推断物种系统发育变得越来越重要。我们开发了一种新工具ComPhy,即“复合距离系统发育”,它基于通过比较基因组对之间的完整基因集计算出的复合距离矩阵来构建原核生物系统发育。
两个基因组之间的复合距离由三个部分定义:基因分散距离(GDD)、基因组断点距离(GBD)和基因含量距离(GCD)。GDD量化直系同源基因沿着基因组坐标从一个基因组到另一个基因组的分散情况;GBD测量两个基因组之间共享的断点;GCD测量两个基因组之间共享直系同源基因的水平。系统发育树使用邻接法从复合距离矩阵构建而成。我们在来自398个完全测序的原核生物基因组的9个数据集上测试了我们的方法。我们的方法构建的树与基于伯杰氏分类法构建的树在四重奏拓扑结构上的一致性达到了90%以上。与其他几种系统发育分析方法相比,我们的方法表现出持续更好的性能。
ComPhy是一种用于全基因组推断基因组间进化关系的快速且强大的工具。它可从http://digbio.missouri.edu/ComPhy下载。