Center for Bioinformatics and Computational Biology, Institute for Advanced Computer Studies and Department of Computer Science, University of Maryland College Park, College Park, MD 20742, USA.
Bioinformatics. 2010 Apr 15;26(8):1057-63. doi: 10.1093/bioinformatics/btq076. Epub 2010 Feb 24.
Understanding the association between genetic diseases and their causal genes is an important problem concerning human health. With the recent influx of high-throughput data describing interactions between gene products, scientists have been provided a new avenue through which these associations can be inferred. Despite the recent interest in this problem, however, there is little understanding of the relative benefits and drawbacks underlying the proposed techniques.
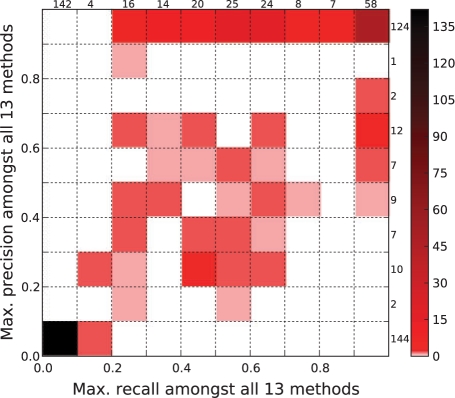
We assessed the utility of physical protein interactions for determining gene-disease associations by examining the performance of seven recently developed computational methods (plus several of their variants). We found that random-walk approaches individually outperform clustering and neighborhood approaches, although most methods make predictions not made by any other method. We show how combining these methods into a consensus method yields Pareto optimal performance. We also quantified how a diffuse topological distribution of disease-related proteins negatively affects prediction quality and are thus able to identify diseases especially amenable to network-based predictions and others for which additional information sources are absolutely required.
The predictions made by each algorithm considered are available online at http://www.cbcb.umd.edu/DiseaseNet.
了解遗传疾病及其因果基因之间的关联是一个关乎人类健康的重要问题。随着高通量数据描述基因产物之间相互作用的不断涌现,科学家们获得了一种新的途径,可以从中推断出这些关联。然而,尽管人们最近对这个问题产生了兴趣,但对于所提出的技术的相对优缺点却知之甚少。
我们通过检查七种最近开发的计算方法(以及它们的几种变体)的性能,评估了物理蛋白质相互作用在确定基因-疾病关联方面的效用。我们发现,随机游走方法单独优于聚类和邻居方法,尽管大多数方法做出了其他方法没有做出的预测。我们展示了如何将这些方法组合成一个共识方法,以获得帕累托最优性能。我们还量化了疾病相关蛋白的弥散拓扑分布如何对预测质量产生负面影响,从而能够识别特别适合基于网络的预测的疾病和其他绝对需要额外信息源的疾病。
每个算法的预测结果都可以在 http://www.cbcb.umd.edu/DiseaseNet 上在线获取。