Department of Electrical and Computer Engineering, Texas A&M University, College Station, Texas 77843-3128, USA.
Curr Genomics. 2009 Sep;10(6):430-45. doi: 10.2174/138920209789177601.
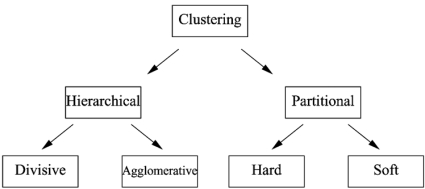
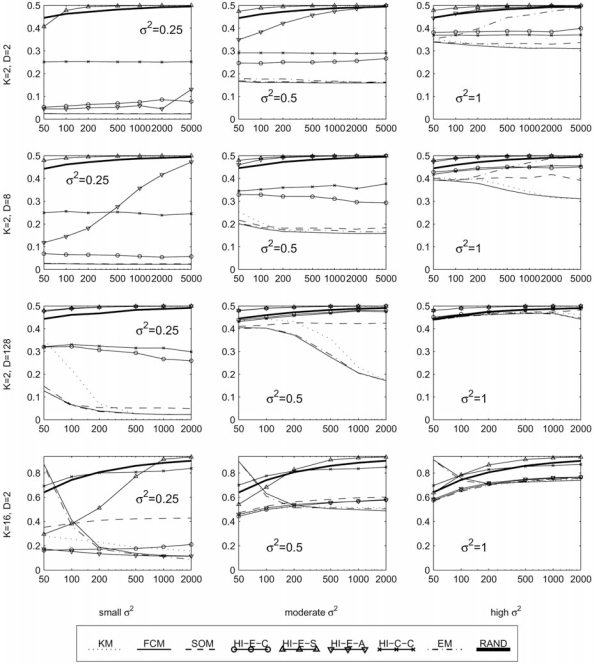
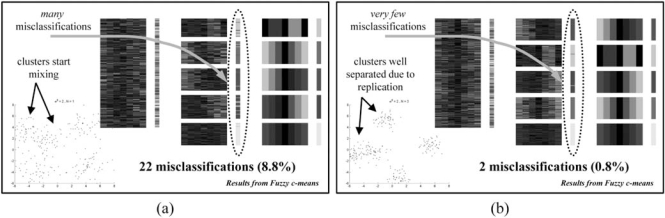
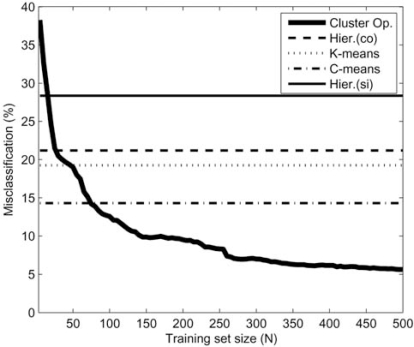
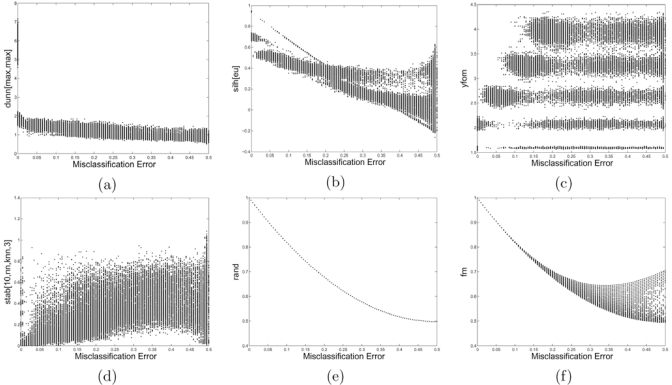
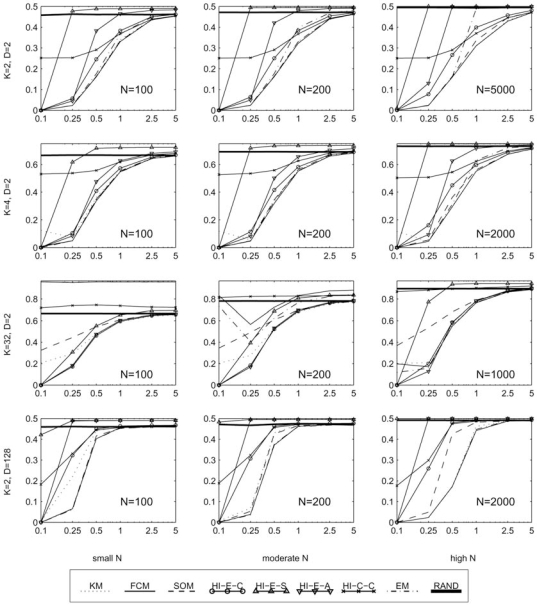
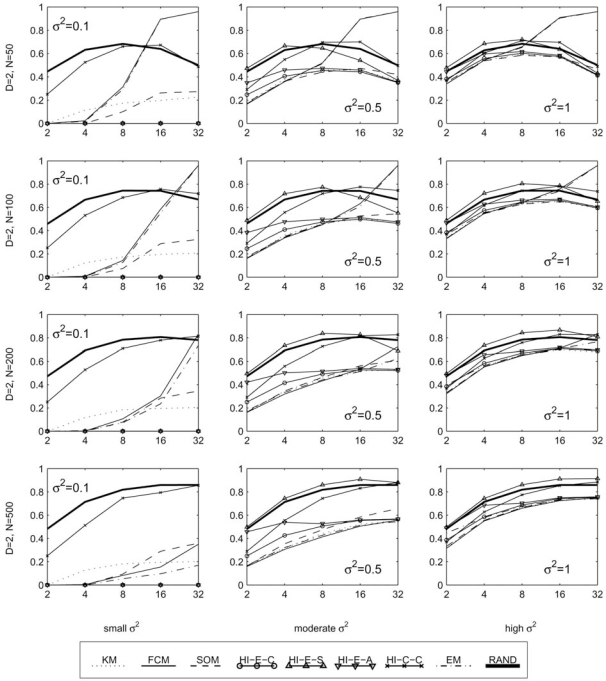
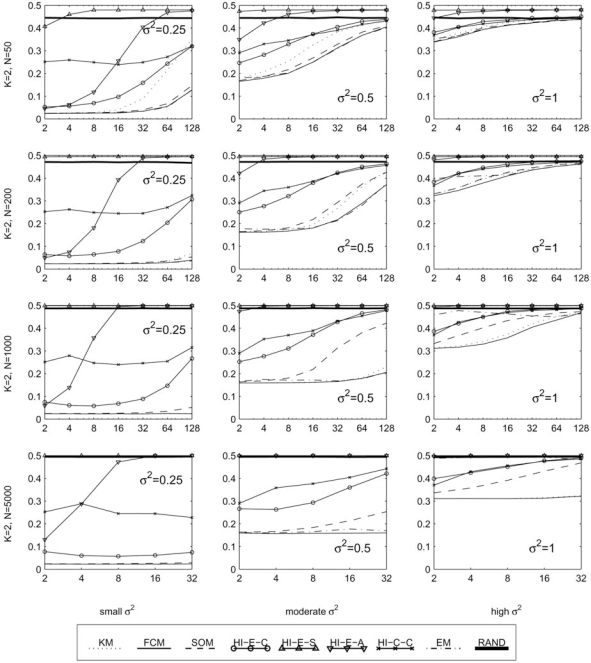
The development of microarray technology has enabled scientists to measure the expression of thousands of genes simultaneously, resulting in a surge of interest in several disciplines throughout biology and medicine. While data clustering has been used for decades in image processing and pattern recognition, in recent years it has joined this wave of activity as a popular technique to analyze microarrays. To illustrate its application to genomics, clustering applied to genes from a set of microarray data groups together those genes whose expression levels exhibit similar behavior throughout the samples, and when applied to samples it offers the potential to discriminate pathologies based on their differential patterns of gene expression. Although clustering has now been used for many years in the context of gene expression microarrays, it has remained highly problematic. The choice of a clustering algorithm and validation index is not a trivial one, more so when applying them to high throughput biological or medical data. Factors to consider when choosing an algorithm include the nature of the application, the characteristics of the objects to be analyzed, the expected number and shape of the clusters, and the complexity of the problem versus computational power available. In some cases a very simple algorithm may be appropriate to tackle a problem, but many situations may require a more complex and powerful algorithm better suited for the job at hand. In this paper, we will cover the theoretical aspects of clustering, including error and learning, followed by an overview of popular clustering algorithms and classical validation indices. We also discuss the relative performance of these algorithms and indices and conclude with examples of the application of clustering to computational biology.
微阵列技术的发展使科学家能够同时测量数千个基因的表达水平,这导致生物学和医学等多个学科对其产生了浓厚的兴趣。虽然数据聚类在图像处理和模式识别中已经使用了几十年,但近年来,它作为一种分析微阵列的流行技术,也加入了这一活动浪潮。为了说明它在基因组学中的应用,聚类应用于一组微阵列数据中的基因,将表达水平在整个样本中表现出相似行为的基因组合在一起,当应用于样本时,它有可能根据基因表达的差异模式来区分病变。尽管聚类在基因表达微阵列的背景下已经使用了多年,但它仍然存在很大的问题。聚类算法和验证指标的选择并不是一件简单的事情,尤其是在将其应用于高通量生物或医学数据时。选择算法时需要考虑的因素包括应用的性质、要分析的对象的特征、预期的聚类数量和形状,以及问题的复杂性与可用的计算能力之间的关系。在某些情况下,一个非常简单的算法可能足以解决问题,但许多情况下可能需要更复杂和强大的算法来更好地处理手头的问题。在本文中,我们将涵盖聚类的理论方面,包括误差和学习,然后概述流行的聚类算法和经典的验证指标。我们还讨论了这些算法和指标的相对性能,并以聚类在计算生物学中的应用为例进行了总结。