Graduate Group in Biophysics, University of California San Francisco, San Francisco, California, United States of America.
PLoS Comput Biol. 2010 Feb 26;6(2):e1000689. doi: 10.1371/journal.pcbi.1000689.
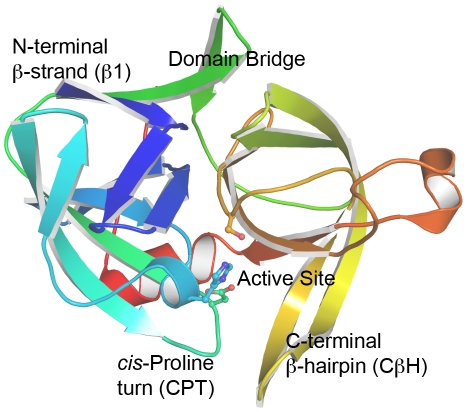
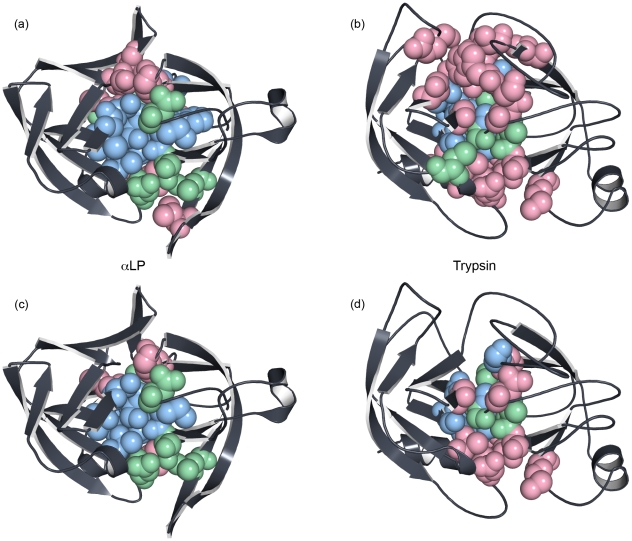
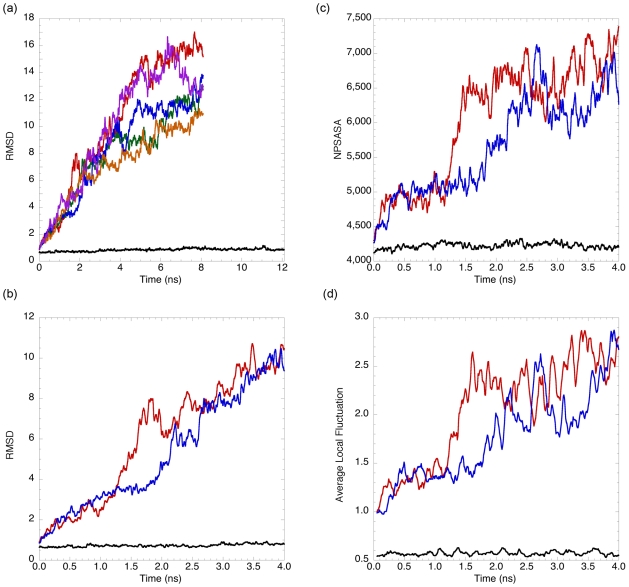
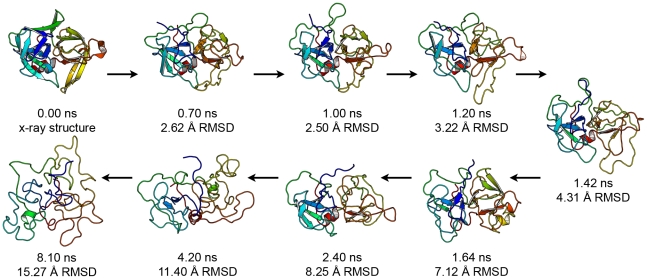
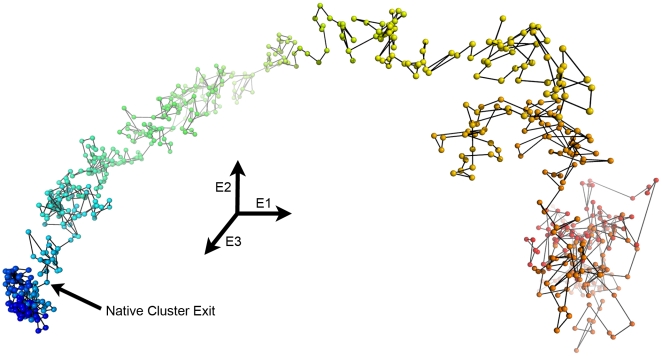
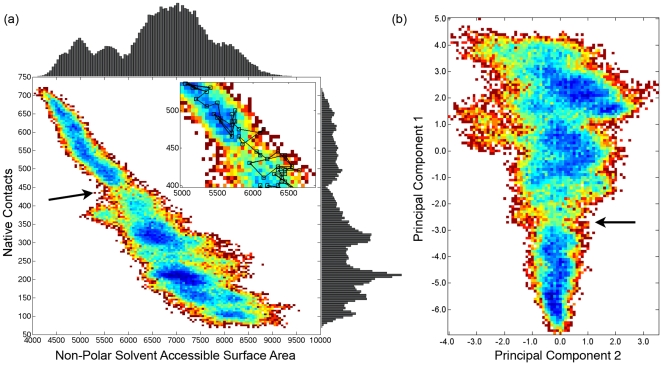
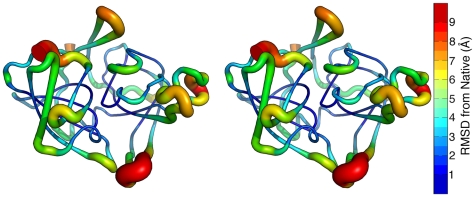
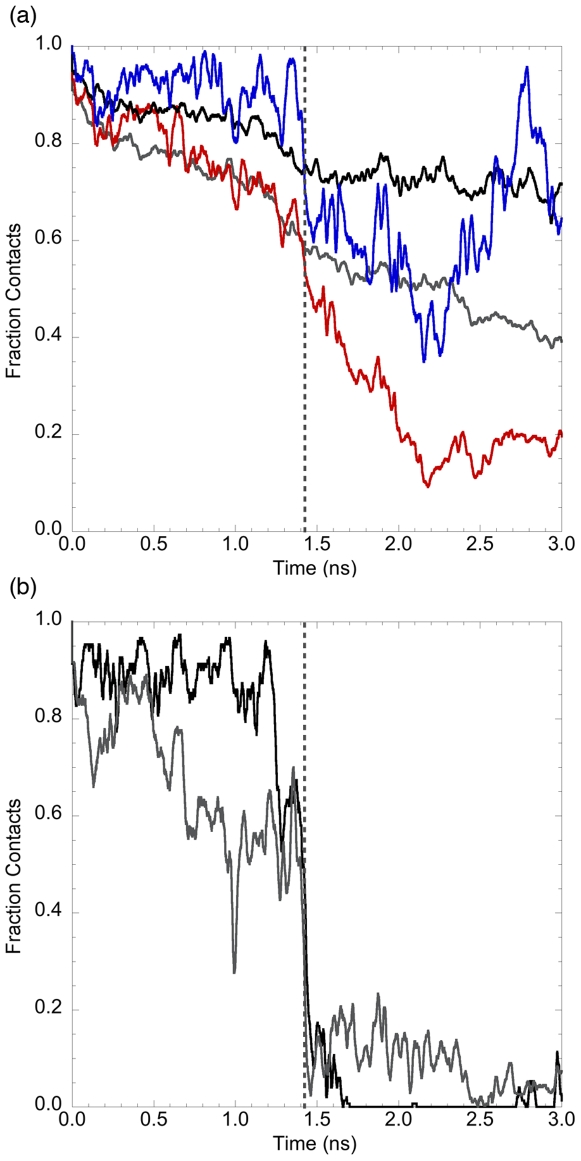
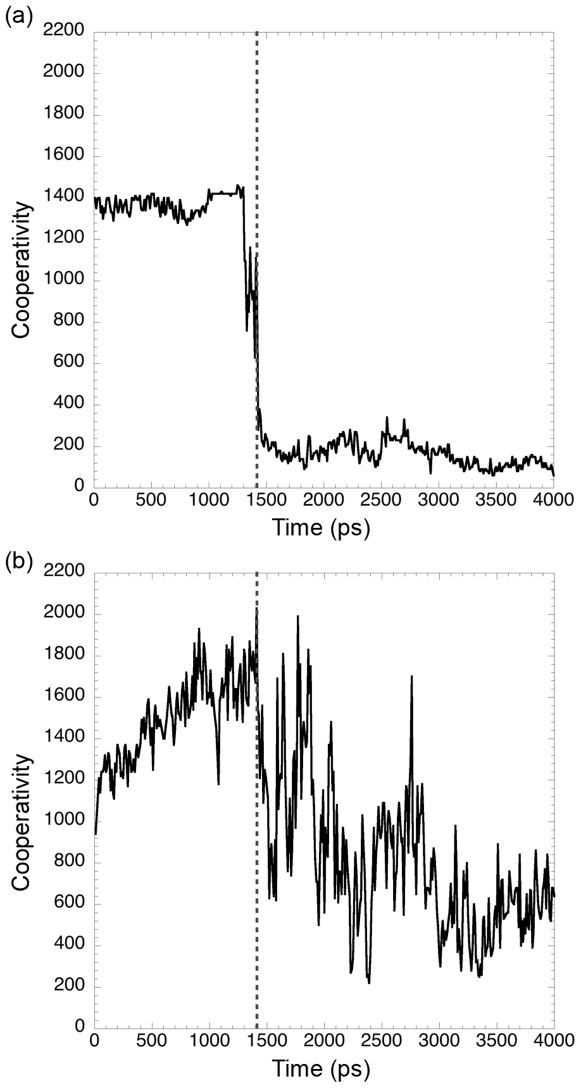
Kinetically stable proteins, those whose stability is derived from their slow unfolding kinetics and not thermodynamics, are examples of evolution's best attempts at suppressing unfolding. Especially in highly proteolytic environments, both partially and fully unfolded proteins face potential inactivation through degradation and/or aggregation, hence, slowing unfolding can greatly extend a protein's functional lifetime. The prokaryotic serine protease alpha-lytic protease (alphaLP) has done just that, as its unfolding is both very slow (t(1/2) approximately 1 year) and so cooperative that partial unfolding is negligible, providing a functional advantage over its thermodynamically stable homologs, such as trypsin. Previous studies have identified regions of the domain interface as critical to alphaLP unfolding, though a complete description of the unfolding pathway is missing. In order to identify the alphaLP unfolding pathway and the mechanism for its extreme cooperativity, we performed high temperature molecular dynamics unfolding simulations of both alphaLP and trypsin. The simulated alphaLP unfolding pathway produces a robust transition state ensemble consistent with prior biochemical experiments and clearly shows that unfolding proceeds through a preferential disruption of the domain interface. Through a novel method of calculating unfolding cooperativity, we show that alphaLP unfolds extremely cooperatively while trypsin unfolds gradually. Finally, by examining the behavior of both domain interfaces, we propose a model for the differential unfolding cooperativity of alphaLP and trypsin involving three key regions that differ between the kinetically stable and thermodynamically stable classes of serine proteases.
动力学稳定的蛋白质,其稳定性来自于缓慢的展开动力学而不是热力学,是进化在抑制展开方面的最佳尝试的例子。特别是在高度蛋白水解的环境中,部分和完全展开的蛋白质都面临着通过降解和/或聚集而潜在失活的风险,因此,减缓展开可以大大延长蛋白质的功能寿命。原核丝氨酸蛋白酶α-溶菌酶(αLP)就是这样做的,因为它的展开既非常缓慢(t(1/2)约为 1 年),又非常协作,以至于部分展开可以忽略不计,这为其提供了相对于热力学稳定的同源物(如胰蛋白酶)的功能优势。先前的研究已经确定了结构域界面的区域对αLP 展开至关重要,尽管仍缺少对展开途径的完整描述。为了确定αLP 的展开途径及其极端协作的机制,我们对αLP 和胰蛋白酶进行了高温分子动力学展开模拟。模拟的αLP 展开途径产生了一个稳健的过渡态集合,与先前的生化实验一致,并清楚地表明展开是通过优先破坏结构域界面进行的。通过一种计算展开协同性的新方法,我们表明αLP 展开具有极高的协同性,而胰蛋白酶则逐渐展开。最后,通过检查两个结构域界面的行为,我们提出了一个关于αLP 和胰蛋白酶的差异展开协同性的模型,涉及到动力学稳定和热力学稳定的丝氨酸蛋白酶类之间的三个关键区域的差异。