Institute of Ecology and Evolution, University of Bern, 3012 Bern, Switzerland.
BMC Bioinformatics. 2010 Mar 4;11:116. doi: 10.1186/1471-2105-11-116.
The estimation of demographic parameters from genetic data often requires the computation of likelihoods. However, the likelihood function is computationally intractable for many realistic evolutionary models, and the use of Bayesian inference has therefore been limited to very simple models. The situation changed recently with the advent of Approximate Bayesian Computation (ABC) algorithms allowing one to obtain parameter posterior distributions based on simulations not requiring likelihood computations.
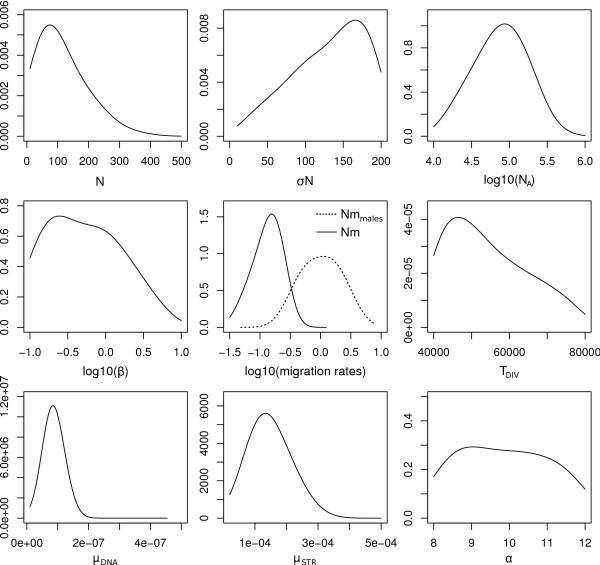
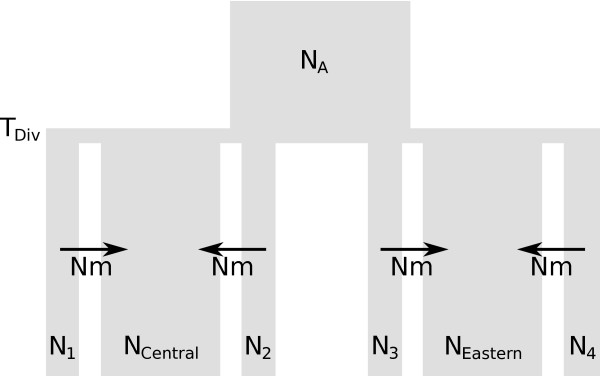
Here we present ABCtoolbox, a series of open source programs to perform Approximate Bayesian Computations (ABC). It implements various ABC algorithms including rejection sampling, MCMC without likelihood, a Particle-based sampler and ABC-GLM. ABCtoolbox is bundled with, but not limited to, a program that allows parameter inference in a population genetics context and the simultaneous use of different types of markers with different ploidy levels. In addition, ABCtoolbox can also interact with most simulation and summary statistics computation programs. The usability of the ABCtoolbox is demonstrated by inferring the evolutionary history of two evolutionary lineages of Microtus arvalis. Using nuclear microsatellites and mitochondrial sequence data in the same estimation procedure enabled us to infer sex-specific population sizes and migration rates and to find that males show smaller population sizes but much higher levels of migration than females.
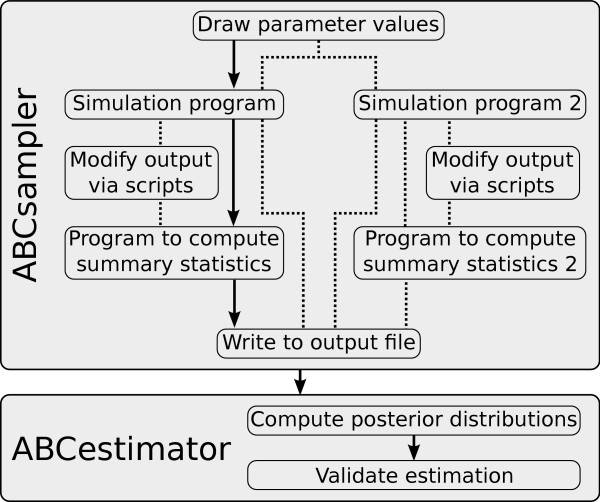
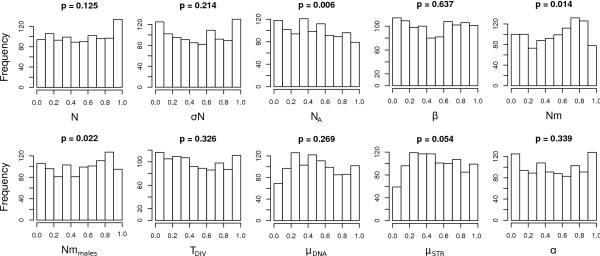
ABCtoolbox allows a user to perform all the necessary steps of a full ABC analysis, from parameter sampling from prior distributions, data simulations, computation of summary statistics, estimation of posterior distributions, model choice, validation of the estimation procedure, and visualization of the results.
从遗传数据估计人口统计学参数通常需要计算似然。然而,对于许多现实的进化模型,似然函数在计算上是难以处理的,因此贝叶斯推断的使用仅限于非常简单的模型。最近,随着近似贝叶斯计算 (ABC) 算法的出现,情况发生了变化,这些算法允许根据不需要似然计算的模拟获得参数后验分布。
这里我们提出了 ABCtoolbox,这是一系列用于执行近似贝叶斯计算 (ABC) 的开源程序。它实现了各种 ABC 算法,包括拒绝采样、无似然的 MCMC、基于粒子的采样器和 ABC-GLM。ABCtoolbox 捆绑了(但不限于)一个程序,该程序允许在群体遗传学背景下进行参数推断,并同时使用具有不同ploidy 水平的不同类型的标记。此外,ABCtoolbox 还可以与大多数模拟和汇总统计计算程序交互。通过推断 Microtus arvalis 的两个进化谱系的进化历史,展示了 ABCtoolbox 的可用性。在相同的估计过程中使用核微卫星和线粒体序列数据,使我们能够推断出性别特异性种群大小和迁移率,并发现雄性的种群大小比雌性小,但迁移水平高得多。
ABCtoolbox 允许用户执行完整的 ABC 分析的所有必要步骤,从参数从先验分布抽样、数据模拟、汇总统计计算、后验分布估计、模型选择、估计过程验证到结果可视化。