UMR7206 Eco-anthropologie, CNRS, Muséum National d'Histoire Naturelle, Université de Paris, Paris, France.
Sub-department of Human Evolution, Department of Organismal Biology, Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden.
Mol Ecol Resour. 2021 May;21(4):1098-1117. doi: 10.1111/1755-0998.13325. Epub 2021 Feb 26.
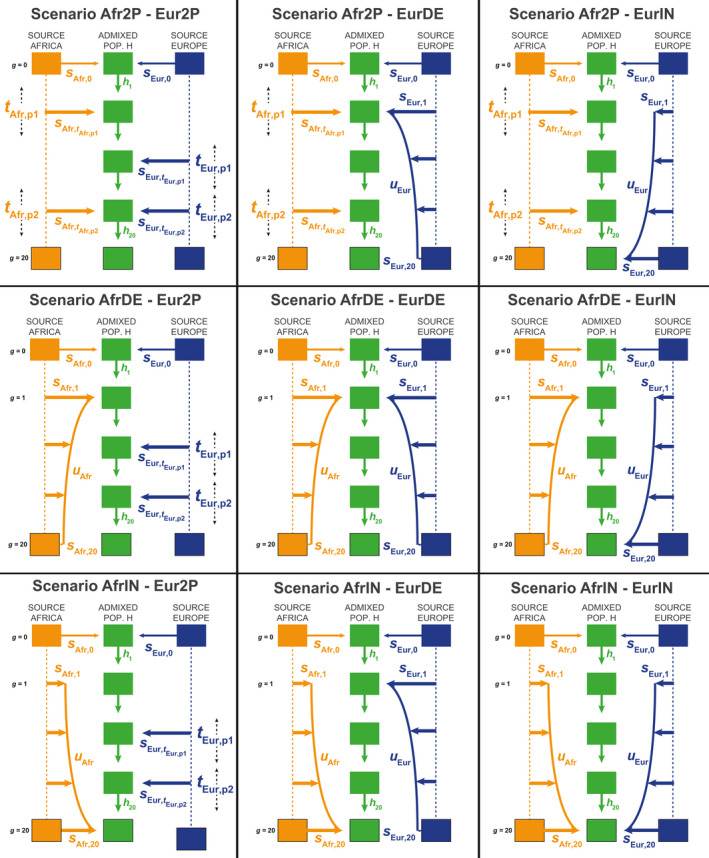
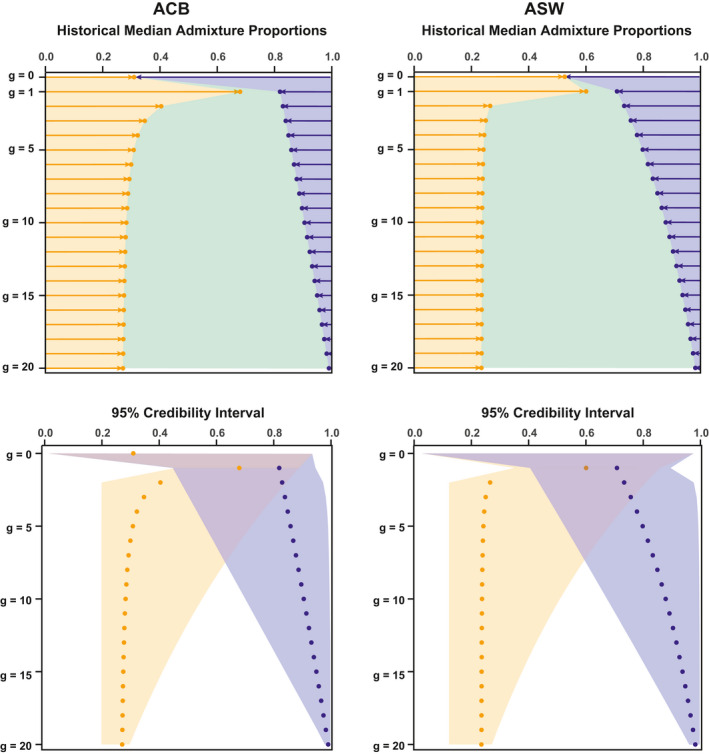
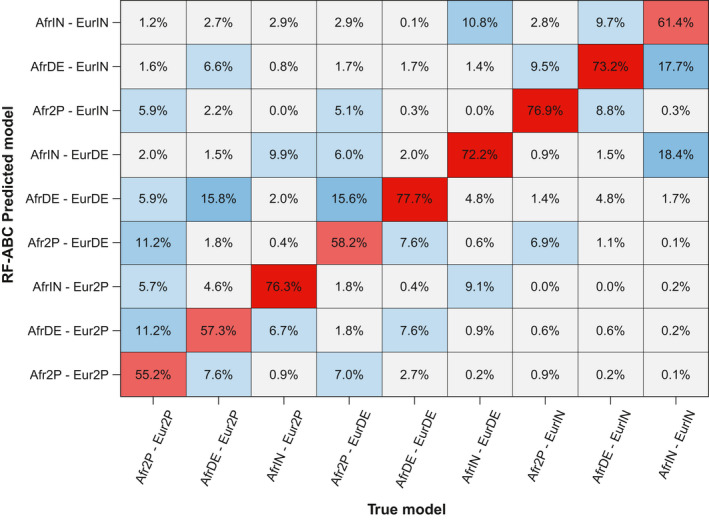
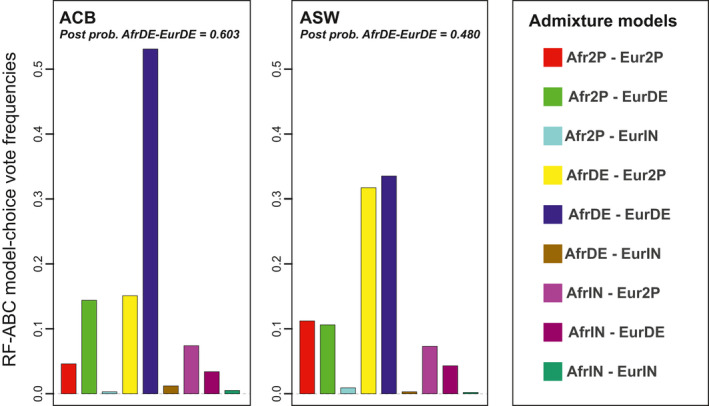
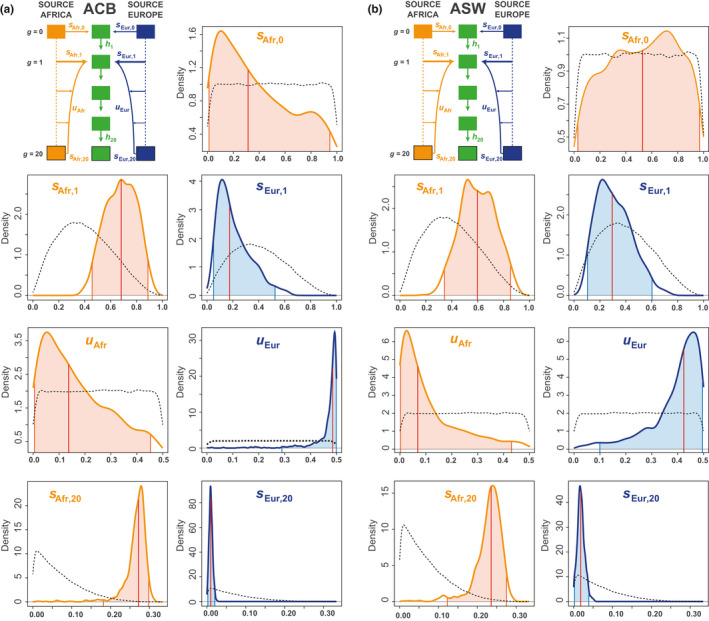
Admixture is a fundamental evolutionary process that has influenced genetic patterns in numerous species. Maximum-likelihood approaches based on allele frequencies and linkage-disequilibrium have been extensively used to infer admixture processes from genome-wide data sets, mostly in human populations. Nevertheless, complex admixture histories, beyond one or two pulses of admixture, remain methodologically challenging to reconstruct. We developed an Approximate Bayesian Computation (ABC) framework to reconstruct highly complex admixture histories from independent genetic markers. We built the software package MetHis to simulate independent SNPs or microsatellites in a two-way admixed population for scenarios with multiple admixture pulses, monotonically decreasing or increasing recurring admixture, or combinations of these scenarios. MetHis allows users to draw model-parameter values from prior distributions set by the user, and, for each simulation, MetHis can calculate numerous summary statistics describing genetic diversity patterns and moments of the distribution of individual admixture fractions. We coupled MetHis with existing machine-learning ABC algorithms and investigated the admixture history of admixed populations. Results showed that random forest ABC scenario-choice could accurately distinguish among most complex admixture scenarios, and errors were mainly found in regions of the parameter space where scenarios were highly nested, and, thus, biologically similar. We focused on African American and Barbadian populations as two study-cases. We found that neural network ABC posterior parameter estimation was accurate and reasonably conservative under complex admixture scenarios. For both admixed populations, we found that monotonically decreasing contributions over time, from Europe and Africa, explained the observed data more accurately than multiple admixture pulses. This approach will allow for reconstructing detailed admixture histories when maximum-likelihood methods are intractable.
混合是一个基本的进化过程,它影响了许多物种的遗传模式。基于等位基因频率和连锁不平衡的最大似然方法已被广泛用于从全基因组数据集推断混合过程,主要是在人类群体中。然而,对于超出一个或两个混合脉冲的复杂混合历史,仍然在方法上难以重建。我们开发了一种近似贝叶斯计算(ABC)框架,用于从独立的遗传标记重建高度复杂的混合历史。我们构建了软件包 MetHis,用于模拟具有多个混合脉冲、单调递减或递增重复混合或这些场景组合的双向混合群体中的独立 SNPs 或微卫星。MetHis 允许用户从用户设置的先验分布中抽取模型参数值,并且对于每个模拟,MetHis 可以计算许多描述遗传多样性模式和个体混合分数分布的矩的汇总统计信息。我们将 MetHis 与现有的机器学习 ABC 算法结合起来,研究了混合群体的混合历史。结果表明,随机森林 ABC 情景选择可以准确地区分大多数复杂的混合情景,并且错误主要出现在情景高度嵌套的参数空间区域,因此在生物学上是相似的。我们以非裔美国人和巴巴多斯人作为两个研究案例。我们发现,在复杂的混合情景下,神经网络 ABC 后验参数估计是准确和合理保守的。对于这两个混合群体,我们发现,从欧洲和非洲随时间单调递减的贡献比多个混合脉冲更准确地解释了观察到的数据。当最大似然方法难以处理时,这种方法将允许重建详细的混合历史。