Department of Physics, The College of William and Mary, Williamsburg, VA, USA.
BMC Bioinformatics. 2010 Apr 8;11:177. doi: 10.1186/1471-2105-11-177.
Time-of-flight mass spectrometry (TOF-MS) has the potential to provide non-invasive, high-throughput screening for cancers and other serious diseases via detection of protein biomarkers in blood or other accessible biologic samples. Unfortunately, this potential has largely been unrealized to date due to the high variability of measurements, uncertainties in the distribution of proteins in a given population, and the difficulty of extracting repeatable diagnostic markers using current statistical tools. With studies consisting of perhaps only dozens of samples, and possibly hundreds of variables, overfitting is a serious complication. To overcome these difficulties, we have developed a Bayesian inductive method which uses model-independent methods of discovering relationships between spectral features. This method appears to efficiently discover network models which not only identify connections between the disease and key features, but also organizes relationships between features--and furthermore creates a stable classifier that categorizes new data at predicted error rates.
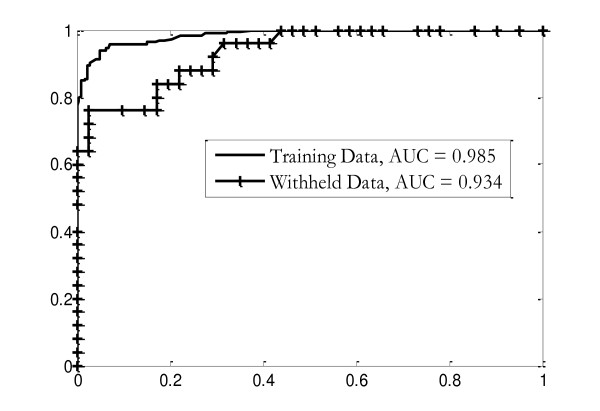
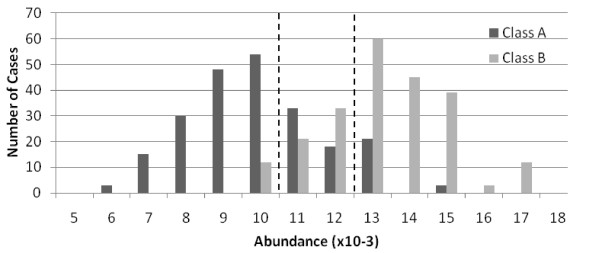
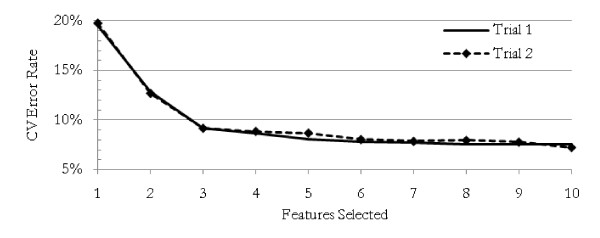
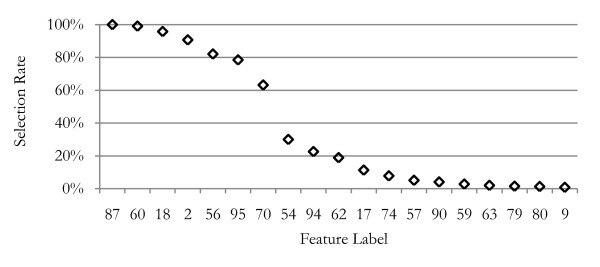
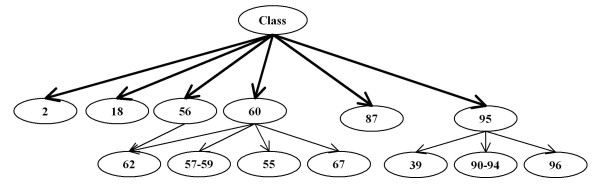
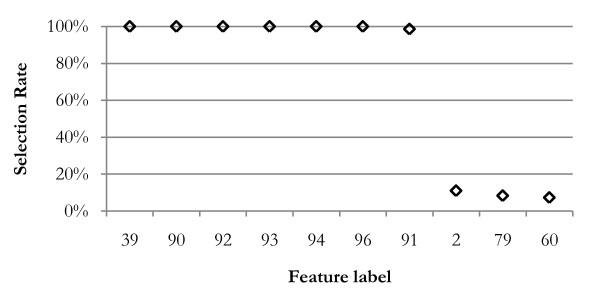
The method was applied to artificial data with known feature relationships and typical TOF-MS variability introduced, and was able to recover those relationships nearly perfectly. It was also applied to blood sera data from a 2004 leukemia study, and showed high stability of selected features under cross-validation. Verification of results using withheld data showed excellent predictive power. The method showed improvement over traditional techniques, and naturally incorporated measurement uncertainties. The relationships discovered between features allowed preliminary identification of a protein biomarker which was consistent with other cancer studies and later verified experimentally.
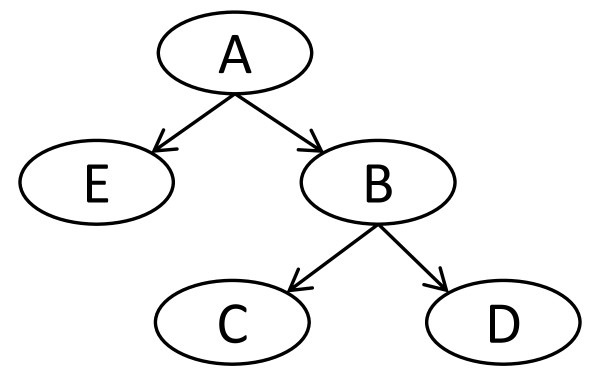
This method appears to avoid overfitting in biologic data and produce stable feature sets in a network model. The network structure provides additional information about the relationships among features that is useful to guide further biochemical analysis. In addition, when used to classify new data, these feature sets are far more consistent than those produced by many traditional techniques.
飞行时间质谱(TOF-MS)有可能通过检测血液或其他可及的生物样本中的蛋白质生物标志物,提供非侵入性、高通量的癌症和其他严重疾病筛查。不幸的是,由于测量的高度可变性、给定人群中蛋白质分布的不确定性以及使用当前统计工具提取可重复诊断标志物的困难,这一潜力迄今在很大程度上尚未实现。由于研究样本数可能只有几十例,甚至数百例,并且可能有数百个变量,因此过拟合是一个严重的问题。为了克服这些困难,我们开发了一种贝叶斯归纳方法,该方法使用独立于模型的方法来发现光谱特征之间的关系。该方法似乎能够有效地发现网络模型,这些模型不仅可以识别疾病与关键特征之间的联系,还可以组织特征之间的关系,并且还可以创建一个稳定的分类器,以预测错误率对新数据进行分类。
该方法应用于具有已知特征关系和典型 TOF-MS 变异性的人工数据,几乎可以完美地恢复这些关系。它还应用于 2004 年白血病研究的血液血清数据,并且在交叉验证下显示出所选特征的高稳定性。使用保留数据进行结果验证表明具有出色的预测能力。该方法显示出优于传统技术的改进,并且自然地包含了测量不确定性。特征之间发现的关系允许初步确定与其他癌症研究一致的蛋白质生物标志物,并随后通过实验验证。
该方法似乎避免了生物数据中的过拟合,并在网络模型中产生了稳定的特征集。网络结构提供了有关特征之间关系的附加信息,这对于指导进一步的生化分析很有用。此外,当用于对新数据进行分类时,这些特征集比许多传统技术生成的特征集更一致。