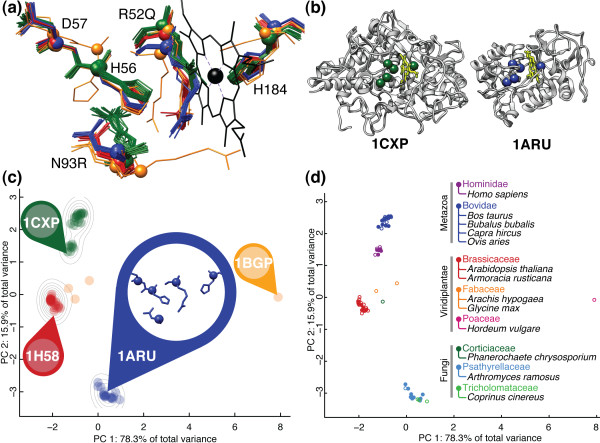
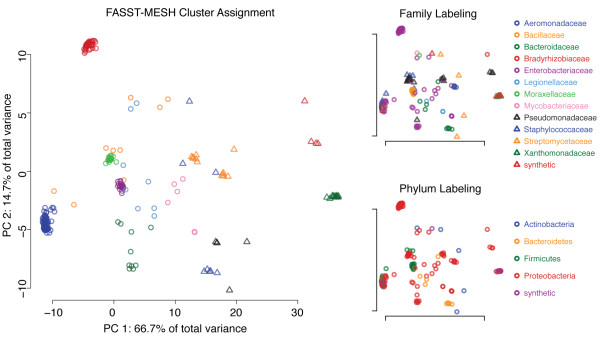
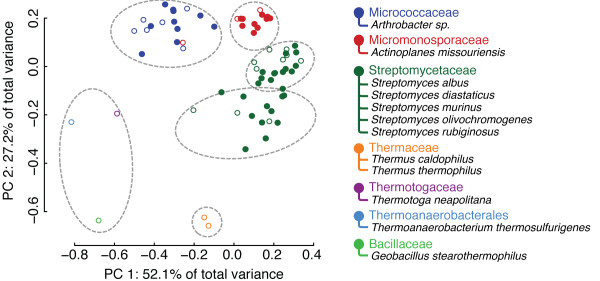
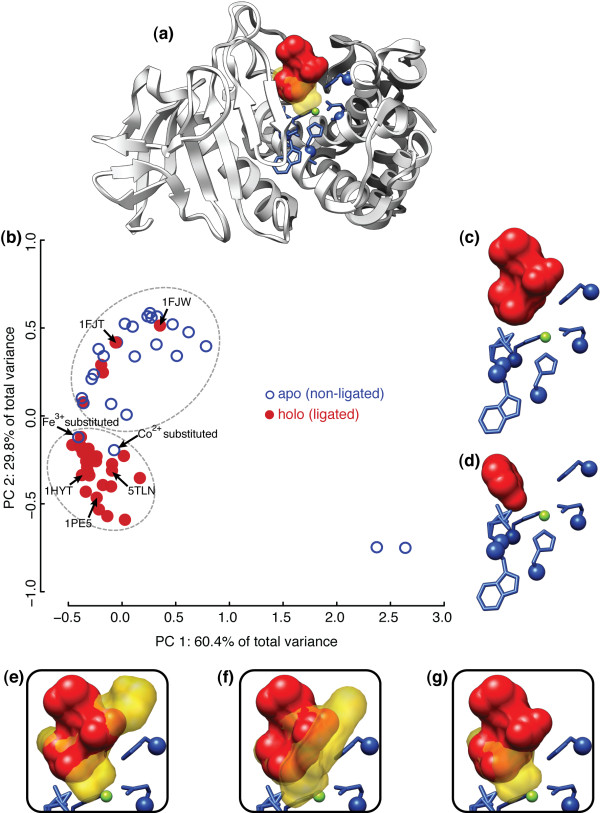
Department of Computer Science, Rice University, Houston, TX, USA.
BMC Bioinformatics. 2010 May 11;11:242. doi: 10.1186/1471-2105-11-242.
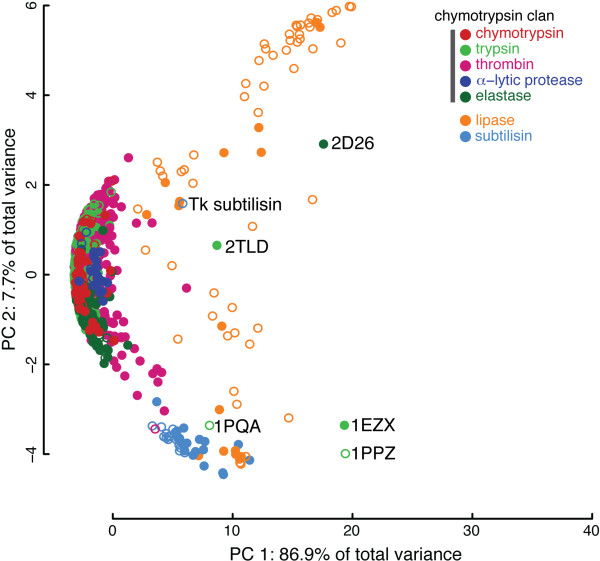
Structural variations caused by a wide range of physico-chemical and biological sources directly influence the function of a protein. For enzymatic proteins, the structure and chemistry of the catalytic binding site residues can be loosely defined as a substructure of the protein. Comparative analysis of drug-receptor substructures across and within species has been used for lead evaluation. Substructure-level similarity between the binding sites of functionally similar proteins has also been used to identify instances of convergent evolution among proteins. In functionally homologous protein families, shared chemistry and geometry at catalytic sites provide a common, local point of comparison among proteins that may differ significantly at the sequence, fold, or domain topology levels.
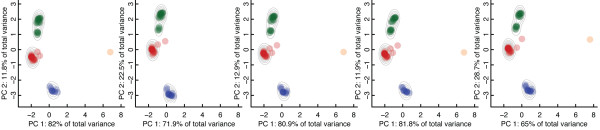
This paper describes two key results that can be used separately or in combination for protein function analysis. The Family-wise Analysis of SubStructural Templates (FASST) method uses all-against-all substructure comparison to determine Substructural Clusters (SCs). SCs characterize the binding site substructural variation within a protein family. In this paper we focus on examples of automatically determined SCs that can be linked to phylogenetic distance between family members, segregation by conformation, and organization by homology among convergent protein lineages. The Motif Ensemble Statistical Hypothesis (MESH) framework constructs a representative motif for each protein cluster among the SCs determined by FASST to build motif ensembles that are shown through a series of function prediction experiments to improve the function prediction power of existing motifs.
FASST contributes a critical feedback and assessment step to existing binding site substructure identification methods and can be used for the thorough investigation of structure-function relationships. The application of MESH allows for an automated, statistically rigorous procedure for incorporating structural variation data into protein function prediction pipelines. Our work provides an unbiased, automated assessment of the structural variability of identified binding site substructures among protein structure families and a technique for exploring the relation of substructural variation to protein function. As available proteomic data continues to expand, the techniques proposed will be indispensable for the large-scale analysis and interpretation of structural data.
由广泛的物理化学和生物来源引起的结构变化直接影响蛋白质的功能。对于酶蛋白,催化结合位点残基的结构和化学性质可以松散地定义为蛋白质的一个亚结构。在物种间和种内对药物受体亚结构进行比较分析已被用于先导评估。功能相似的蛋白质之间结合位点的亚结构相似性也被用于鉴定蛋白质之间趋同进化的实例。在功能同源的蛋白质家族中,催化位点的共享化学和几何形状为蛋白质提供了一个共同的、局部的比较点,这些蛋白质在序列、折叠或结构域拓扑水平上可能有很大的差异。
本文描述了两个可单独使用或组合使用的关键结果,用于蛋白质功能分析。家族内亚结构模板分析(FASST)方法使用全对全亚结构比较来确定亚结构簇(SC)。SC 特征在于蛋白质家族内的结合位点亚结构变化。在本文中,我们重点介绍了自动确定的 SC 的示例,这些示例可以与家族成员之间的系统发育距离、构象分离以及趋同蛋白谱系之间的同源组织相关联。Motif Ensemble Statistical Hypothesis(MESH)框架为 FASST 确定的 SC 中的每个蛋白质簇构建一个代表基序,构建 motif 集合,通过一系列功能预测实验证明,这些集合可以提高现有 motif 的功能预测能力。
FASST 为现有的结合位点亚结构识别方法提供了关键的反馈和评估步骤,并可用于深入研究结构-功能关系。MESH 的应用允许将结构变化数据自动、严格地纳入蛋白质功能预测管道。我们的工作提供了一种对蛋白质结构家族中识别的结合位点亚结构的结构可变性进行无偏、自动评估的方法,以及一种探索亚结构变化与蛋白质功能关系的技术。随着可用蛋白质组数据的不断扩展,所提出的技术对于大规模分析和解释结构数据将是不可或缺的。