Department of Computer Science and Engineering, University of Notre Dame, Notre Dame, IN, USA.
BMC Genomics. 2010 May 17;11:310. doi: 10.1186/1471-2164-11-310.
Several recent studies have demonstrated the use of Roche 454 sequencing technology for de novo transcriptome analysis. Low error rates and high coverage also allow for effective SNP discovery and genetic diversity estimates. However, genetically diverse datasets, such as those sourced from natural populations, pose challenges for assembly programs and subsequent analysis. Further, estimating the effectiveness of transcript discovery using Roche 454 transcriptome data is still a difficult task.
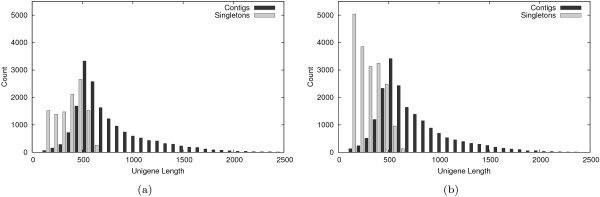
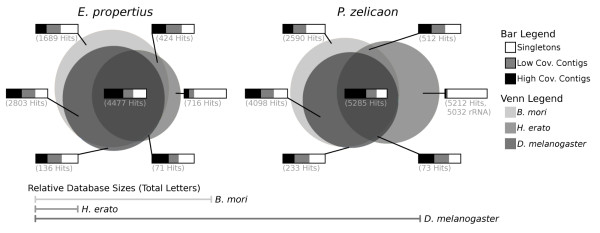
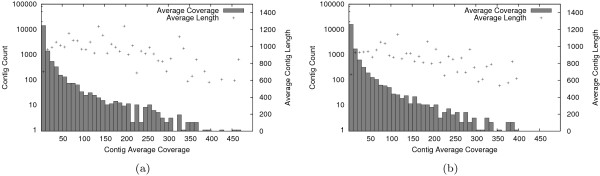
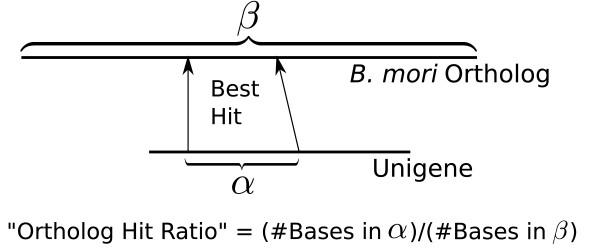
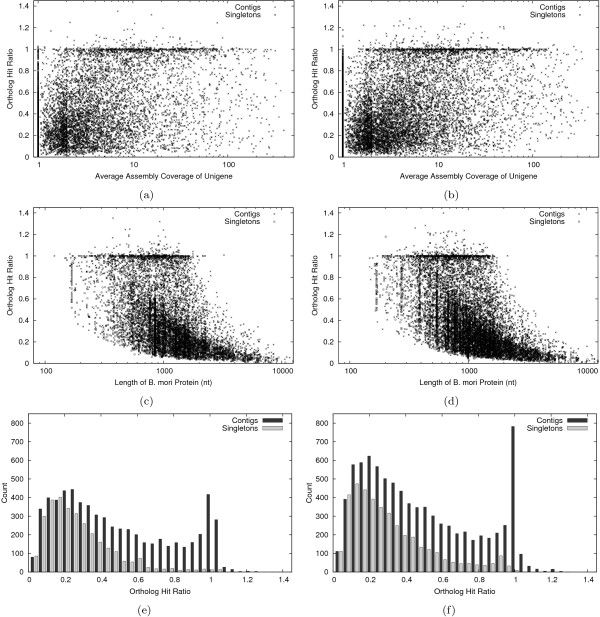
Using the Roche 454 FLX Titanium platform, we sequenced and assembled larval transcriptomes for two butterfly species: the Propertius duskywing, Erynnis propertius (Lepidoptera: Hesperiidae) and the Anise swallowtail, Papilio zelicaon (Lepidoptera: Papilionidae). The Expressed Sequence Tags (ESTs) generated represent a diverse sample drawn from multiple populations, developmental stages, and stress treatments. Despite this diversity, > 95% of the ESTs assembled into long (> 714 bp on average) and highly covered (> 9.6x on average) contigs. To estimate the effectiveness of transcript discovery, we compared the number of bases in the hit region of unigenes (contigs and singletons) to the length of the best match silkworm (Bombyx mori) protein--this "ortholog hit ratio" gives a close estimate on the amount of the transcript discovered relative to a model lepidopteran genome. For each species, we tested two assembly programs and two parameter sets; although CAP3 is commonly used for such data, the assemblies produced by Celera Assembler with modified parameters were chosen over those produced by CAP3 based on contig and singleton counts as well as ortholog hit ratio analysis. In the final assemblies, 1,413 E. propertius and 1,940 P. zelicaon unigenes had a ratio > 0.8; 2,866 E. propertius and 4,015 P. zelicaon unigenes had a ratio > 0.5.
Ultimately, these assemblies and SNP data will be used to generate microarrays for ecoinformatics examining climate change tolerance of different natural populations. These studies will benefit from high quality assemblies with few singletons (less than 26% of bases for each assembled transcriptome are present in unassembled singleton ESTs) and effective transcript discovery (over 6,500 of our putative orthologs cover at least 50% of the corresponding model silkworm gene).
最近的几项研究表明,罗氏 454 测序技术可用于从头转录组分析。低错误率和高覆盖率还允许有效地发现 SNP 和遗传多样性估计。然而,遗传多样性数据集,例如源自自然种群的数据集,给组装程序和后续分析带来了挑战。此外,使用罗氏 454 转录组数据估计转录本发现的有效性仍然是一项艰巨的任务。
我们使用罗氏 454 FLX Titanium 平台对两种蝴蝶物种的幼虫转录组进行了测序和组装:Propertius duskywing,Erynnis propertius(鳞翅目: Hesperiidae)和 Anise swallowtail,Papilio zelicaon(鳞翅目: Papilionidae)。生成的表达序列标签(EST)代表了来自多个种群、发育阶段和应激处理的多种样本。尽管存在这种多样性,但超过 95%的 EST 组装成长度超过 714 bp(平均长度)且高度覆盖(平均覆盖率超过 9.6x)的 contigs。为了估计转录本发现的有效性,我们将 unigenes(contigs 和 singletons)的 hit 区域中的碱基数与最佳匹配家蚕(Bombyx mori)蛋白的长度进行了比较 - 这种“同源 hit 比”可以对相对于模型鳞翅目基因组的转录本发现量进行密切估计。对于每个物种,我们测试了两种组装程序和两种参数集;尽管 CAP3 通常用于此类数据,但基于 contig 和 singleton 计数以及同源 hit 比分析,选择了经过修改参数的 Celera Assembler 生成的组装结果,而不是 CAP3 生成的组装结果。在最终的组装中,1413 个 E. propertius 和 1940 个 P. zelicaon unigenes 的比值> 0.8;2866 个 E. propertius 和 4015 个 P. zelicaon unigenes 的比值> 0.5。
最终,这些组装和 SNP 数据将用于为生态信息学研究生成微阵列,以研究不同自然种群对气候变化的耐受性。这些研究将受益于高质量的组装和较少的 singletons(每个组装转录组中不到 26%的碱基存在于未组装的 singleton EST 中)以及有效的转录本发现(我们的大约 6500 个假定同源物覆盖了至少 50%的对应模型家蚕基因)。