Institut für Botanik III, Heinrich-Heine Universität, Universitätstrasse 1, Düsseldorf, Germany.
Syst Biol. 2010 May;59(3):288-97. doi: 10.1093/sysbio/syq003. Epub 2010 Mar 1.
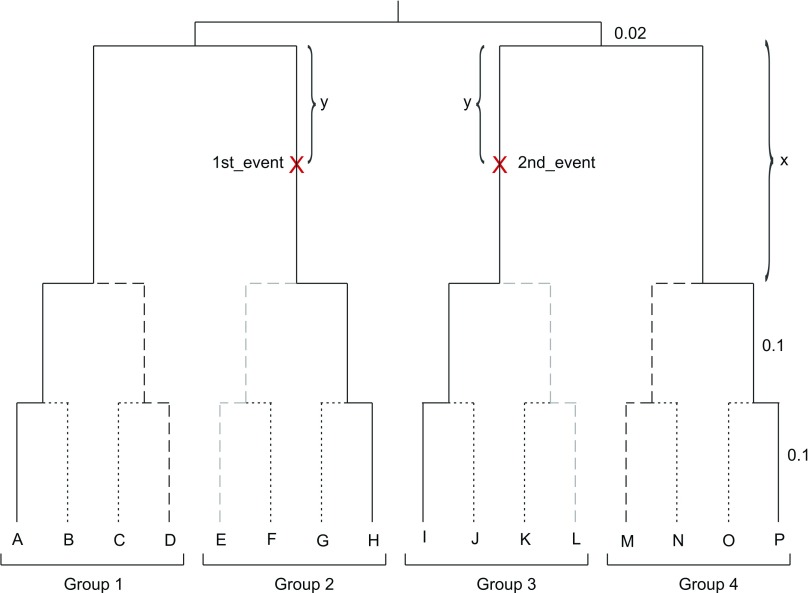
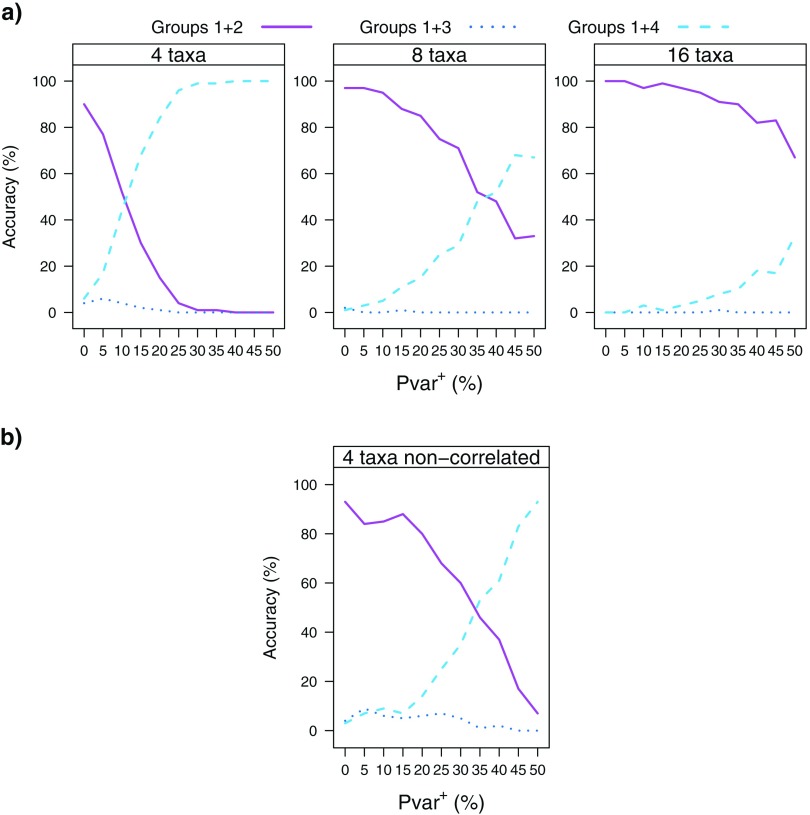
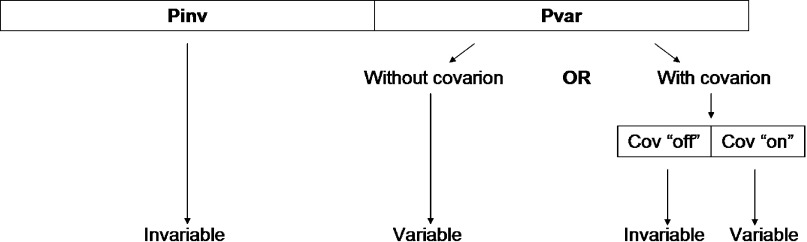
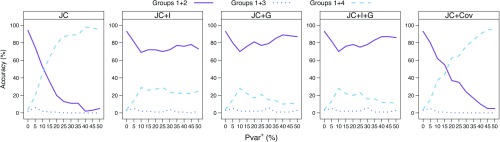
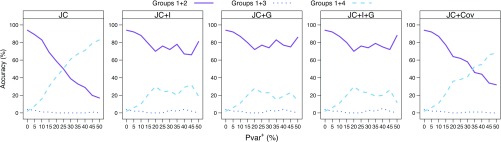
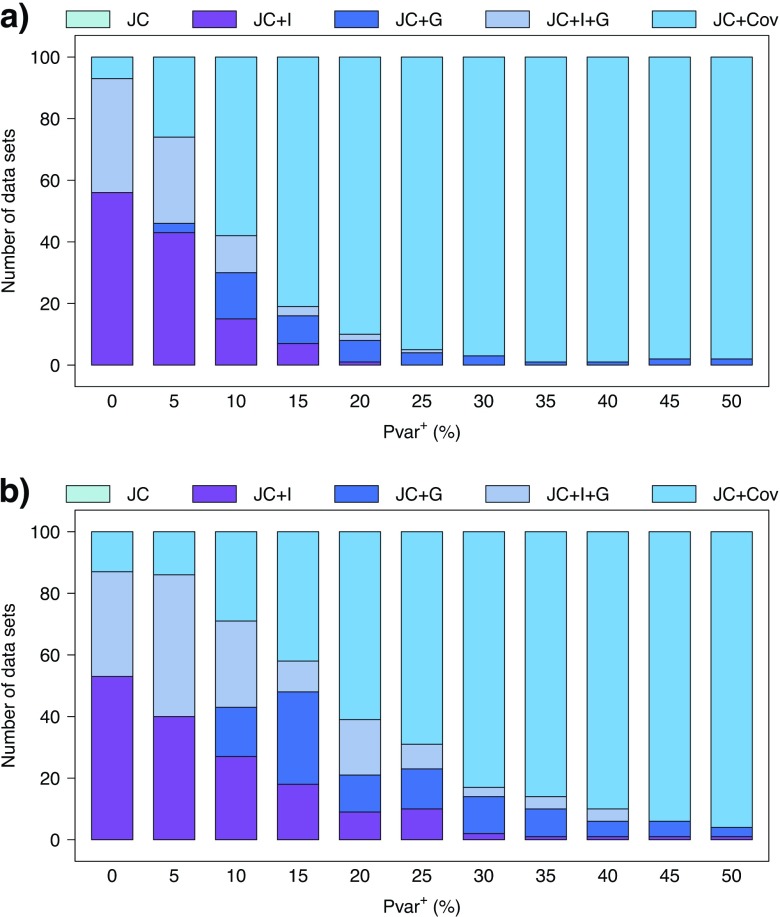
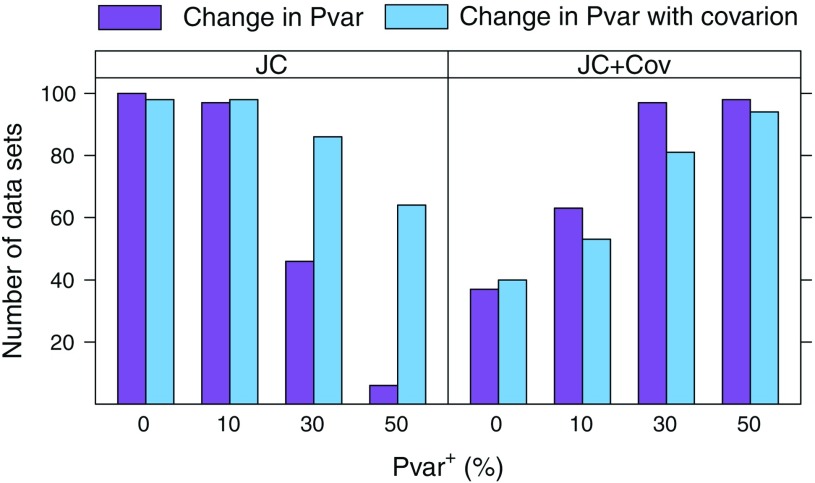
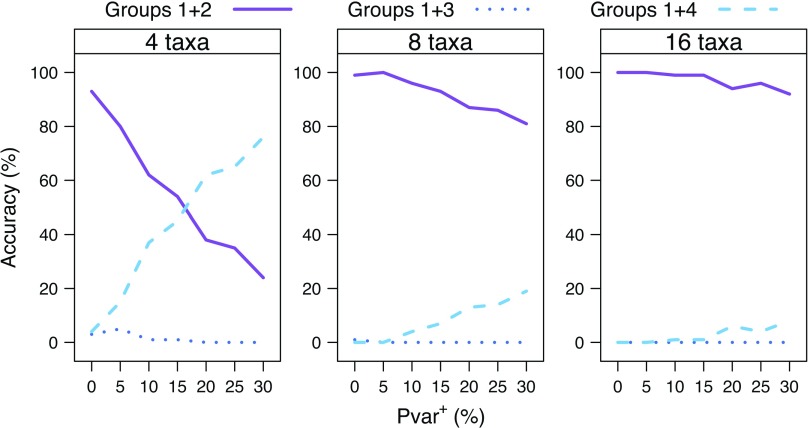
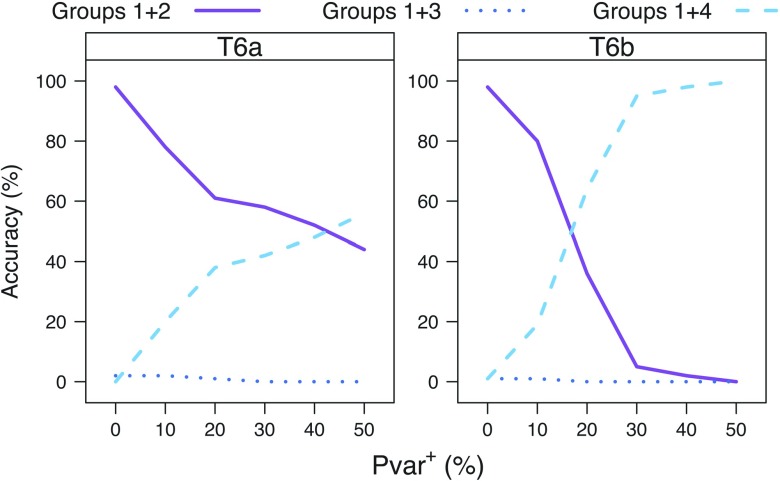
Commonly used phylogenetic models assume a homogeneous process through time in all parts of the tree. However, it is known that these models can be too simplistic as they do not account for nonhomogeneous lineage-specific properties. In particular, it is now widely recognized that as constraints on sequences evolve, the proportion and positions of variable sites can vary between lineages causing heterotachy. The extent to which this model misspecification affects tree reconstruction is still unknown. Here, we evaluate the effect of changes in the proportions and positions of variable sites on model fit and tree estimation. We consider 5 current models of nucleotide sequence evolution in a Bayesian Markov chain Monte Carlo framework as well as maximum parsimony (MP). We show that for a tree with 4 lineages where 2 nonsister taxa undergo a change in the proportion of variable sites tree reconstruction under the best-fitting model, which is chosen using a relative test, often results in the wrong tree. In this case, we found that an absolute test of model fit is a better predictor of tree estimation accuracy. We also found further evidence that MP is not immune to heterotachy. In addition, we show that increased sampling of taxa that have undergone a change in proportion and positions of variable sites is critical for accurate tree reconstruction.
常用的系统发育模型假设树的所有部分在整个时间内都具有均匀的进化过程。然而,已知这些模型可能过于简单化,因为它们没有考虑到非均匀的谱系特异性属性。特别是,现在已经广泛认识到,随着序列约束的进化,变异位点的比例和位置在谱系之间可能会发生变化,导致异速进化。这种模型指定不当对树重建的影响程度尚不清楚。在这里,我们评估了变异位点比例和位置变化对模型拟合和树估计的影响。我们在贝叶斯马尔可夫链蒙特卡罗框架中考虑了 5 种当前的核苷酸序列进化模型,以及最大简约法 (MP)。我们表明,对于一个有 4 个谱系的树,其中 2 个非姐妹分类群的变异位点比例发生变化,使用相对测试选择最佳拟合模型进行树重建,通常会导致错误的树。在这种情况下,我们发现模型拟合的绝对测试是树估计准确性的更好预测指标。我们还进一步发现证据表明,MP 不能免受异速进化的影响。此外,我们表明,增加对比例和位置发生变化的分类群的采样对于准确的树重建至关重要。