Program in Molecular and Computational Biology, University of Southern California, Los Angeles, CA, USA.
Bioinformatics. 2010 Jun 15;26(12):i261-8. doi: 10.1093/bioinformatics/btq201.
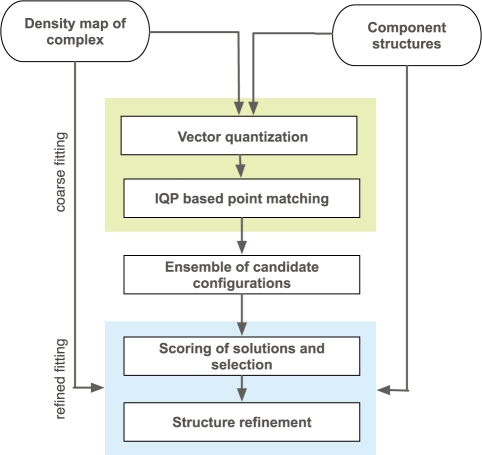
Single-particle cryo electron microscopy (cryoEM) typically produces density maps of macromolecular assemblies at intermediate to low resolution (approximately 5-30 A). By fitting high-resolution structures of assembly components into these maps, pseudo-atomic models can be obtained. Optimizing the quality-of-fit of all components simultaneously is challenging due to the large search space that makes the exhaustive search over all possible component configurations computationally unfeasible.
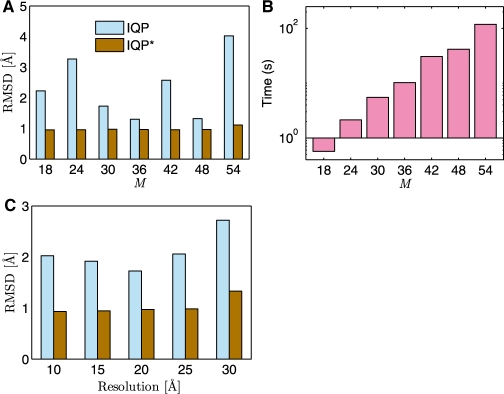
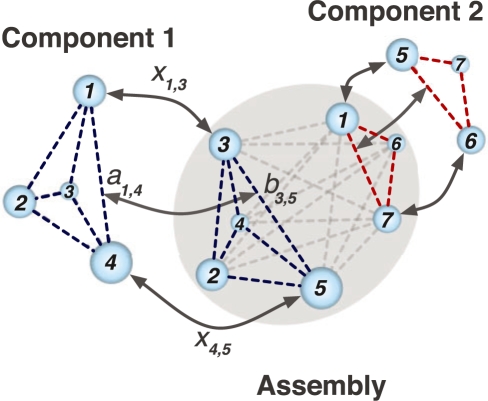
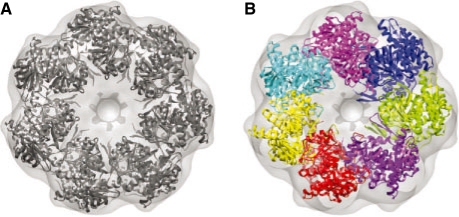
We developed an efficient mathematical programming algorithm that simultaneously fits all component structures into an assembly density map. The fitting is formulated as a point set matching problem involving several point sets that represent component and assembly densities at a reduced complexity level. In contrast to other point matching algorithms, our algorithm is able to match multiple point sets simultaneously and not only based on their geometrical equivalence, but also based on the similarity of the density in the immediate point neighborhood. In addition, we present an efficient refinement method based on the Iterative Closest Point registration algorithm. The integer quadratic programming method generates an assembly configuration in a few seconds. This efficiency allows the generation of an ensemble of candidate solutions that can be assessed by an independent scoring function. We benchmarked the method using simulated density maps of 11 protein assemblies at 20 A, and an experimental cryoEM map at 23.5 A resolution. Our method was able to generate assembly structures with root-mean-square errors <6.5 A, which have been further reduced to <1.8 A by the local refinement procedure.
The program is available upon request as a Matlab code package.
Supplementary data are available at Bioinformatics Online.
单颗粒低温电子显微镜(cryoEM)通常可以在中低分辨率(约 5-30Å)下获得大分子组装体的密度图。通过将组装体组件的高分辨率结构拟合到这些图谱中,可以获得伪原子模型。由于搜索空间很大,使得对所有可能的组件配置进行穷举搜索在计算上不可行,因此同时优化所有组件的拟合质量具有挑战性。
我们开发了一种有效的数学规划算法,该算法可同时将所有组件结构拟合到组装密度图中。拟合被表述为涉及几个点集的点集匹配问题,这些点集以降低的复杂度级别表示组件和组装密度。与其他点匹配算法不同,我们的算法能够同时匹配多个点集,并且不仅基于它们的几何等价性,还基于紧邻点邻域中密度的相似性。此外,我们提出了一种基于迭代最近点注册算法的高效细化方法。整数二次规划方法在几秒钟内生成一个组装配置。这种效率允许生成一组候选解决方案,这些候选解决方案可以通过独立的评分函数进行评估。我们使用 11 个蛋白质组装体在 20Å 的模拟密度图和 23.5Å 的实验 cryoEM 图进行了方法的基准测试。我们的方法能够生成均方根误差<6.5Å 的组装结构,通过局部细化过程进一步将其降低到<1.8Å。
该程序可根据要求作为 Matlab 代码包提供。
补充数据可在“Bioinformatics Online”上获得。