Toyota Technological Institute, Chicago, IL 60637, USA.
Bioinformatics. 2010 Jun 15;26(12):i310-7. doi: 10.1093/bioinformatics/btq193.
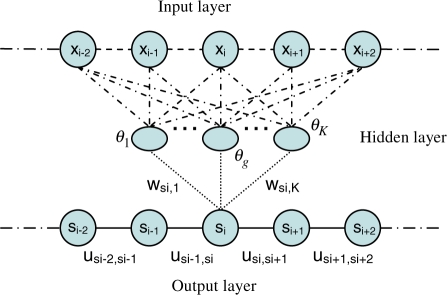
One of the major bottlenecks with ab initio protein folding is an effective conformation sampling algorithm that can generate native-like conformations quickly. The popular fragment assembly method generates conformations by restricting the local conformations of a protein to short structural fragments in the PDB. This method may limit conformations to a subspace to which the native fold does not belong because (i) a protein with really new fold may contain some structural fragments not in the PDB and (ii) the discrete nature of fragments may prevent them from building a native-like fold. Previously we have developed a conditional random fields (CRF) method for fragment-free protein folding that can sample conformations in a continuous space and demonstrated that this CRF method compares favorably to the popular fragment assembly method. However, the CRF method is still limited by its capability of generating conformations compatible with a sequence.
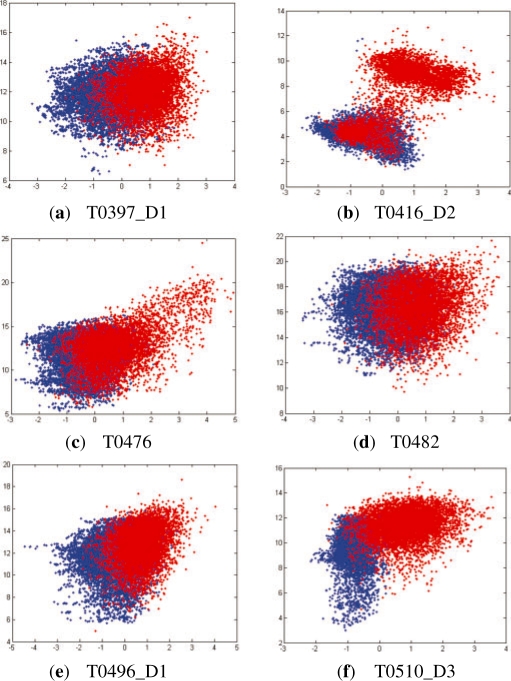
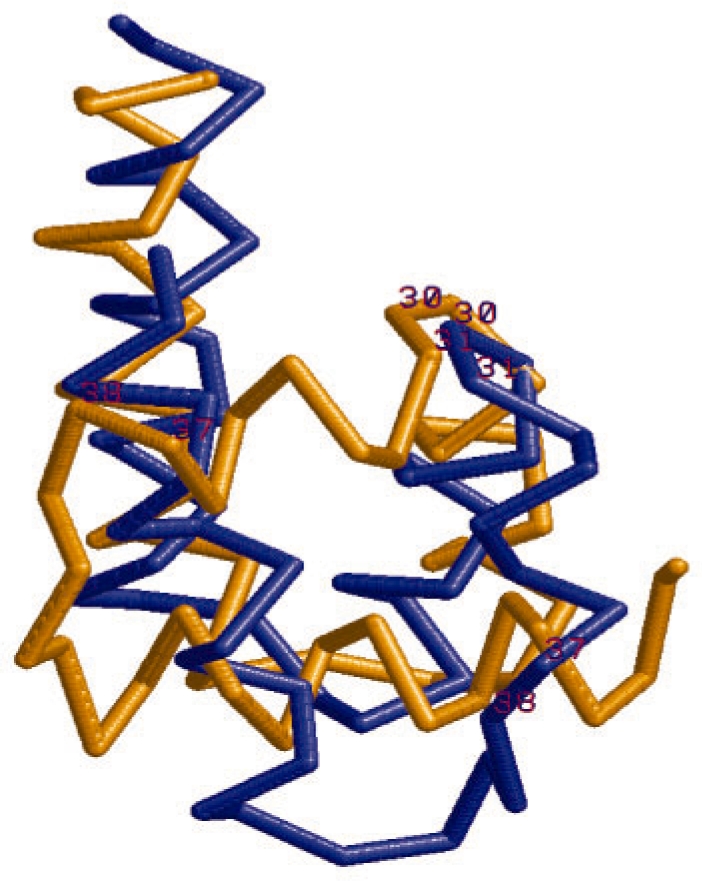
We present a new fragment-free approach to protein folding using a recently invented probabilistic graphical model conditional neural fields (CNF). This new CNF method is much more powerful than CRF in modeling the sophisticated protein sequence-structure relationship and thus, enables us to generate native-like conformations more easily. We show that when coupled with a simple energy function and replica exchange Monte Carlo simulation, our CNF method can generate decoys much better than CRF on a variety of test proteins including the CASP8 free-modeling targets. In particular, our CNF method can predict a correct fold for T0496_D1, one of the two CASP8 targets with truly new fold. Our predicted model for T0496 is significantly better than all the CASP8 models.
从头开始蛋白质折叠的主要瓶颈之一是一种有效的构象采样算法,该算法可以快速生成类似天然的构象。流行的片段组装方法通过将蛋白质的局部构象限制在 PDB 中的短结构片段来生成构象。由于以下原因,该方法可能将构象限制在天然折叠不属于的子空间中:(i)具有真正新折叠的蛋白质可能包含一些不在 PDB 中的结构片段,(ii)片段的离散性质可能阻止它们构建类似天然的折叠。之前,我们已经开发了一种无片段的蛋白质折叠条件随机场(CRF)方法,可以在连续空间中采样构象,并证明该 CRF 方法与流行的片段组装方法相比具有优势。然而,CRF 方法仍然受到其生成与序列兼容的构象的能力的限制。
我们提出了一种使用最近发明的概率图形模型条件神经网络场(CNF)的无片段蛋白质折叠新方法。与 CRF 相比,这种新的 CNF 方法在建模复杂的蛋白质序列-结构关系方面具有更强的能力,因此使我们更容易生成类似天然的构象。我们表明,当与简单的能量函数和 replica 交换蒙特卡罗模拟结合使用时,我们的 CNF 方法可以在各种测试蛋白质(包括 CASP8 自由建模靶标)上比 CRF 更好地生成诱饵。特别是,我们的 CNF 方法可以预测 T0496_D1 的正确折叠,这是两个具有真正新折叠的 CASP8 靶标之一。我们对 T0496 的预测模型明显优于所有 CASP8 模型。