Laboratory of Systems Pharmacology, Harvard Medical School, Boston, MA 02115, USA; Department of Systems Biology, Harvard Medical School, Boston, MA 02115, USA.
Cell Syst. 2019 Apr 24;8(4):292-301.e3. doi: 10.1016/j.cels.2019.03.006. Epub 2019 Apr 17.
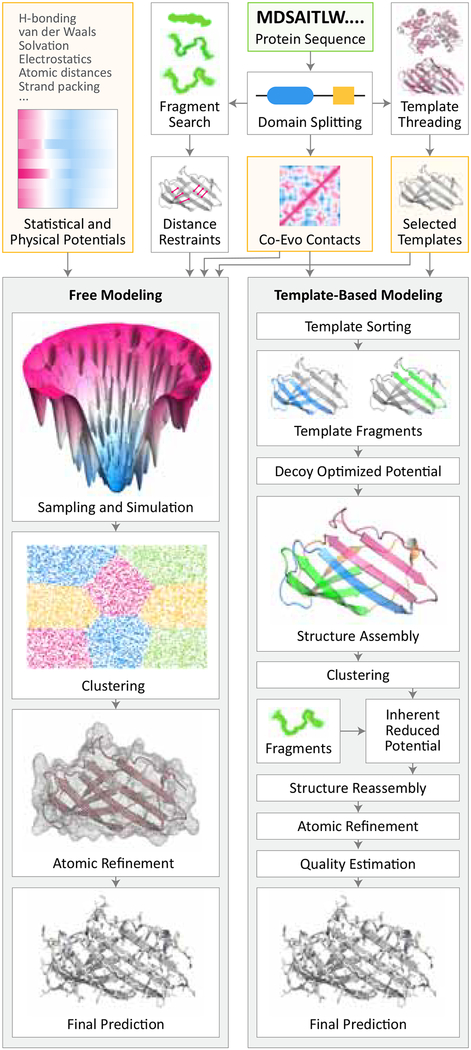
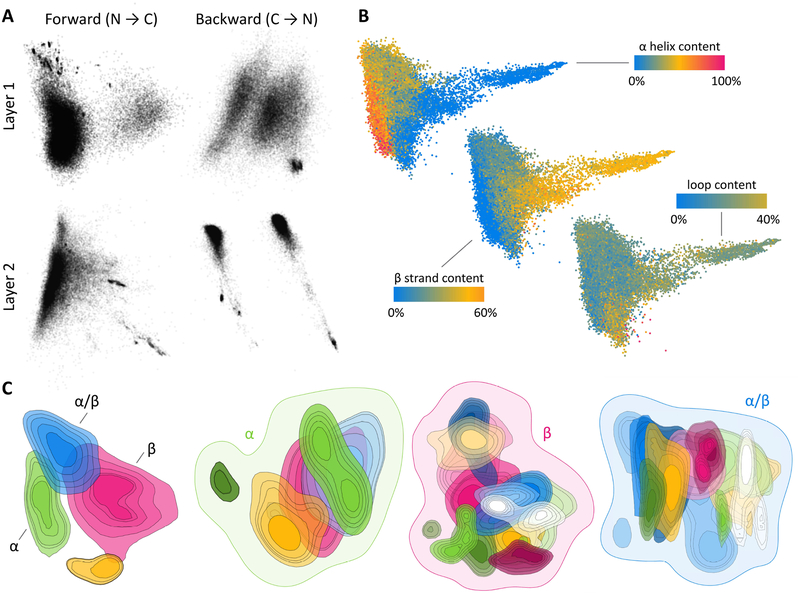
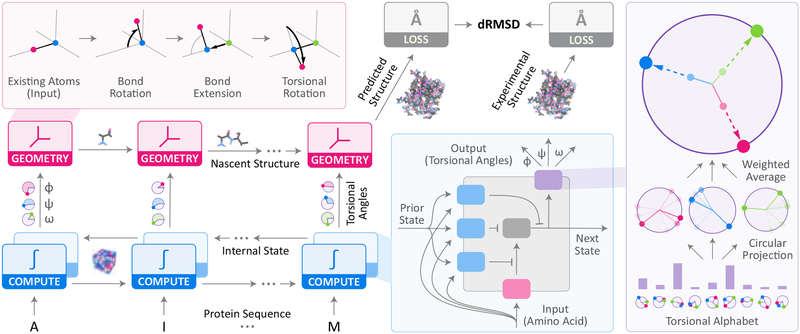
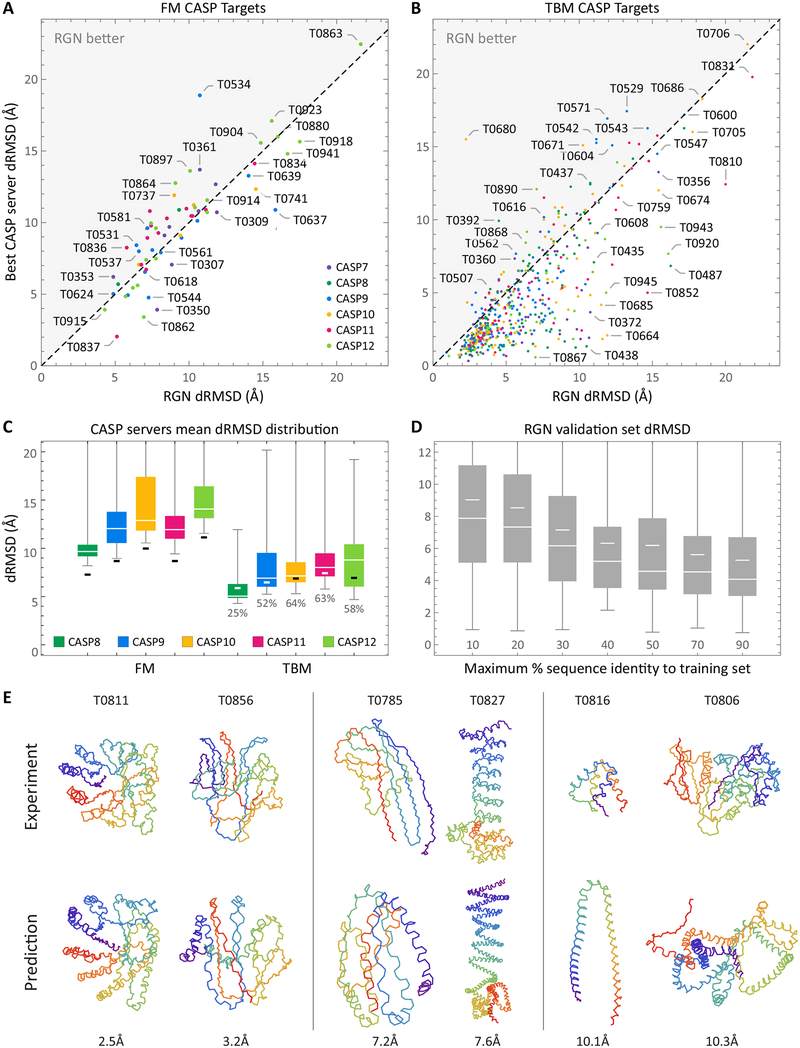
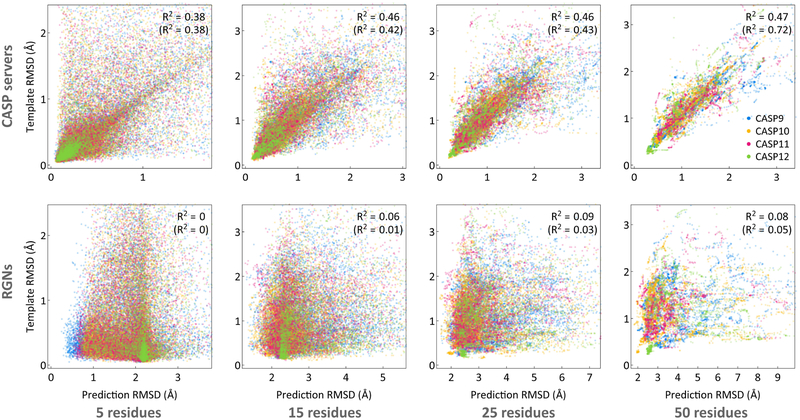
Predicting protein structure from sequence is a central challenge of biochemistry. Co-evolution methods show promise, but an explicit sequence-to-structure map remains elusive. Advances in deep learning that replace complex, human-designed pipelines with differentiable models optimized end to end suggest the potential benefits of similarly reformulating structure prediction. Here, we introduce an end-to-end differentiable model for protein structure learning. The model couples local and global protein structure via geometric units that optimize global geometry without violating local covalent chemistry. We test our model using two challenging tasks: predicting novel folds without co-evolutionary data and predicting known folds without structural templates. In the first task, the model achieves state-of-the-art accuracy, and in the second, it comes within 1-2 Å; competing methods using co-evolution and experimental templates have been refined over many years, and it is likely that the differentiable approach has substantial room for further improvement, with applications ranging from drug discovery to protein design.
从序列预测蛋白质结构是生物化学的核心挑战。共进化方法显示出前景,但明确的序列到结构的映射仍然难以捉摸。深度学习的进步用可微分的模型替代了复杂的、人工设计的流水线,并进行端到端优化,这表明类似地重新制定结构预测具有潜在的好处。在这里,我们引入了一个用于蛋白质结构学习的端到端可微分模型。该模型通过几何单元来耦合局部和全局蛋白质结构,这些几何单元在不违反局部共价化学的情况下优化全局几何形状。我们使用两个具有挑战性的任务来测试我们的模型:在没有共进化数据的情况下预测新的折叠结构,以及在没有结构模板的情况下预测已知的折叠结构。在第一个任务中,该模型达到了最先进的准确性,在第二个任务中,它的误差在 1-2Å 以内;使用共进化和实验模板的竞争方法已经经过多年的改进,因此可微分方法很可能还有很大的改进空间,其应用范围从药物发现到蛋白质设计。