Department of Computer Science and Engineering, University of Washington, Seattle, WA, USA.
Bioinformatics. 2010 Jun 15;26(12):i334-42. doi: 10.1093/bioinformatics/btq175.
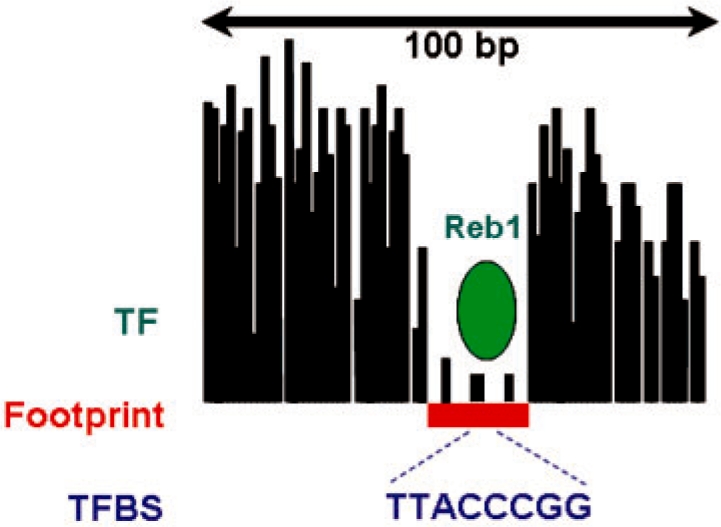
A global map of transcription factor binding sites (TFBSs) is critical to understanding gene regulation and genome function. DNaseI digestion of chromatin coupled with massively parallel sequencing (digital genomic footprinting) enables the identification of protein-binding footprints with high resolution on a genome-wide scale. However, accurately inferring the locations of these footprints remains a challenging computational problem.
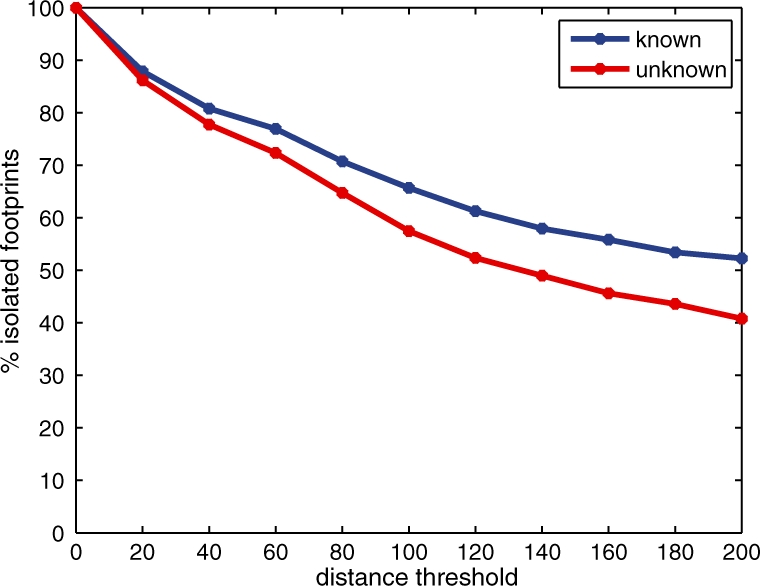
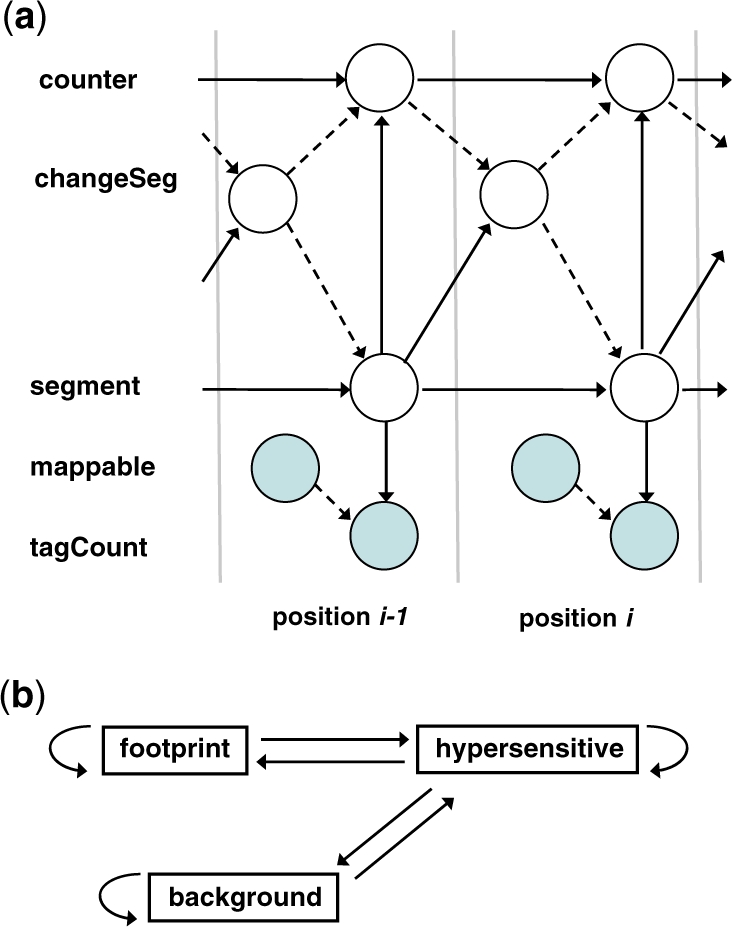
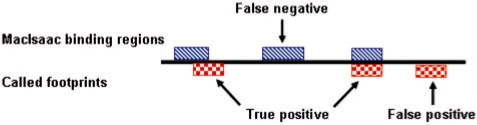
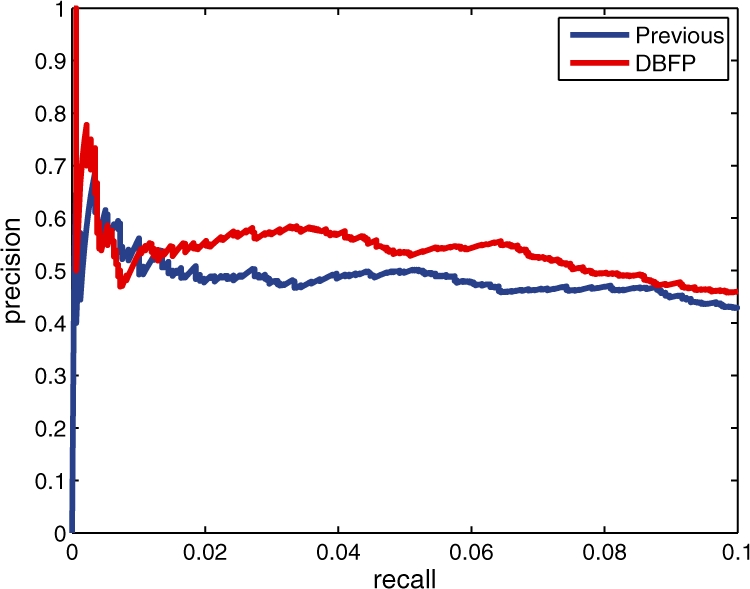
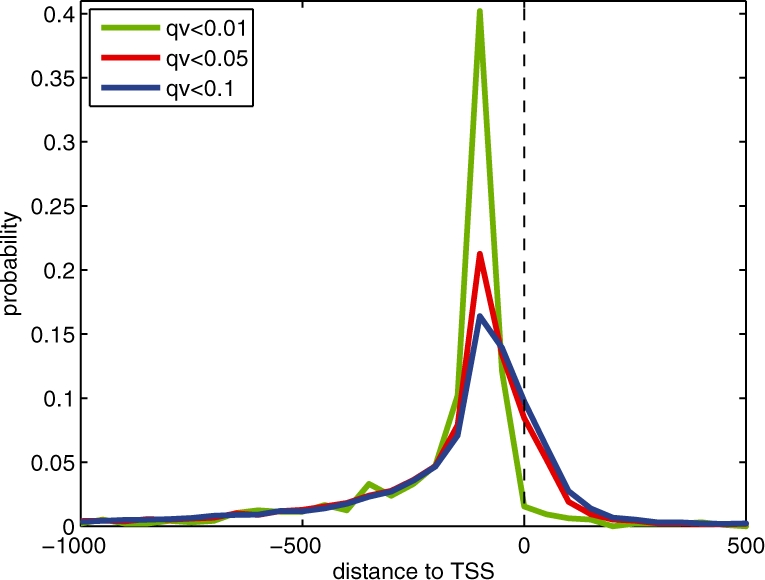
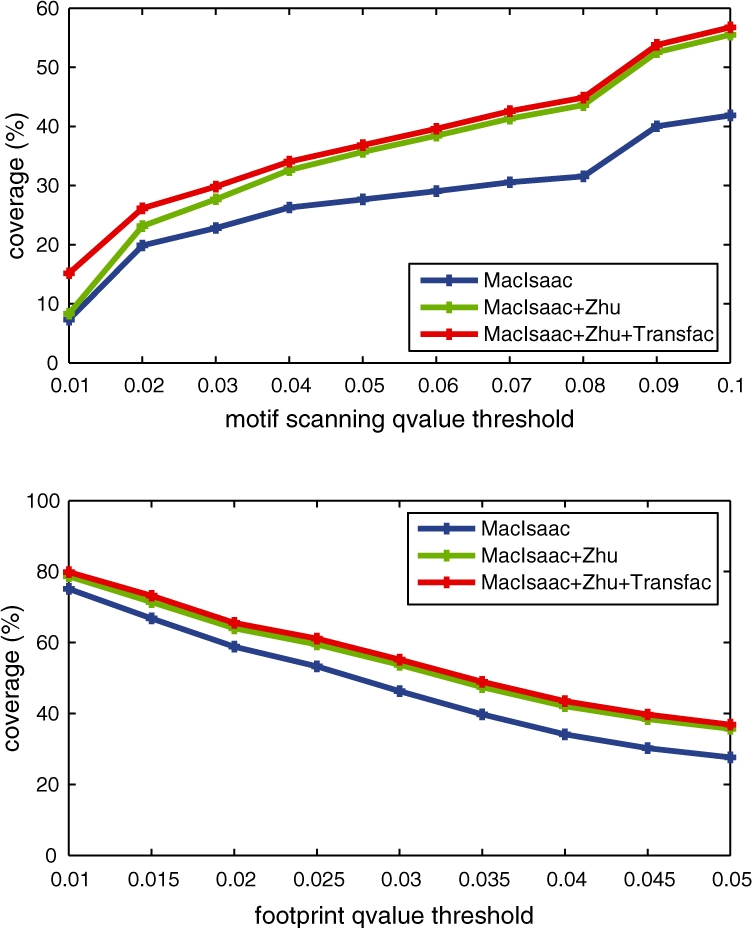
We present a dynamic Bayesian network-based approach for the identification and assignment of statistical confidence estimates to protein-binding footprints from digital genomic footprinting data. The method, DBFP, allows footprints to be identified in a probabilistic framework and outperforms our previously described algorithm in terms of precision at a fixed recall. Applied to a digital footprinting data set from Saccharomyces cerevisiae, DBFP identifies 4679 statistically significant footprints within intergenic regions. These footprints are mainly located near transcription start sites and are strongly enriched for known TFBSs. Footprints containing no known motif are preferentially located proximal to other footprints, consistent with cooperative binding of these footprints. DBFP also identifies a set of statistically significant footprints in the yeast coding regions. Many of these footprints coincide with the boundaries of antisense transcripts, and the most significant footprints are enriched for binding sites of the chromatin-associated factors Abf1 and Rap1.
Supplementary material is available at Bioinformatics online.
转录因子结合位点(TFBS)的全球图谱对于理解基因调控和基因组功能至关重要。用 DNA 酶 I 消化染色质,再结合大规模平行测序(数字基因组足迹法),可以在全基因组范围内以高分辨率识别蛋白质结合的足迹。然而,准确推断这些足迹的位置仍然是一个具有挑战性的计算问题。
我们提出了一种基于动态贝叶斯网络的方法,用于从数字基因组足迹数据中识别和分配蛋白质结合足迹的统计置信度估计值。该方法 DBFP 允许在概率框架中识别足迹,并在固定召回率下优于我们之前描述的算法在精度方面的表现。将该方法应用于来自酿酒酵母的数字足迹数据集,DBFP 在基因间区域内识别出 4679 个具有统计学意义的足迹。这些足迹主要位于转录起始位点附近,并且强烈富集已知的 TFBS。没有已知基序的足迹优先位于其他足迹附近,这与这些足迹的协同结合一致。DBFP 还在酵母编码区域中识别出一组具有统计学意义的足迹。这些足迹中的许多与反义转录本的边界重合,而最显著的足迹则富集了与染色质相关因子 Abf1 和 Rap1 结合的位点。
补充材料可在“Bioinformatics”在线获取。