Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Cambridge, CB10 1SA, UK.
Bioinformatics. 2010 Jun 15;26(12):i367-73. doi: 10.1093/bioinformatics/btq217.
Sequence assembly is a difficult problem whose importance has grown again recently as the cost of sequencing has dramatically dropped. Most new sequence assembly software has started by building a de Bruijn graph, avoiding the overlap-based methods used previously because of the computational cost and complexity of these with very large numbers of short reads. Here, we show how to use suffix array-based methods that have formed the basis of recent very fast sequence mapping algorithms to find overlaps and generate assembly string graphs asymptotically faster than previously described algorithms.
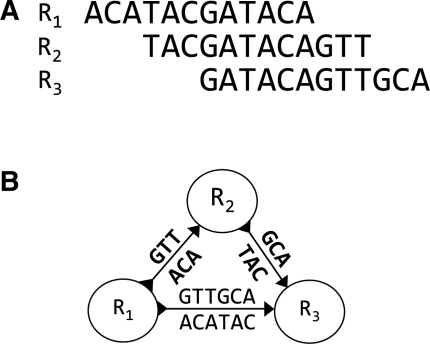
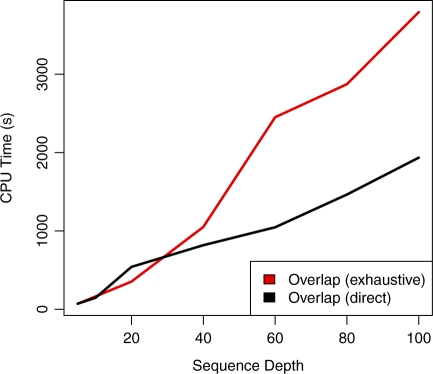
Standard overlap assembly methods have time complexity O(N(2)), where N is the sum of the lengths of the reads. We use the Ferragina-Manzini index (FM-index) derived from the Burrows-Wheeler transform to find overlaps of length at least tau among a set of reads. As well as an approach that finds all overlaps then implements transitive reduction to produce a string graph, we show how to output directly only the irreducible overlaps, significantly shrinking memory requirements and reducing compute time to O(N), independent of depth. Overlap-based assembly methods naturally handle mixed length read sets, including capillary reads or long reads promised by the third generation sequencing technologies. The algorithms we present here pave the way for overlap-based assembly approaches to be developed that scale to whole vertebrate genome de novo assembly.
序列组装是一个难题,随着测序成本的大幅降低,其重要性再次凸显。大多数新的序列组装软件都是从构建 de Bruijn 图开始的,因为以前使用的基于重叠的方法由于计算成本和非常大量的短读的复杂性而被回避。在这里,我们展示了如何使用后缀数组方法,这些方法已成为最近非常快速的序列映射算法的基础,以比以前描述的算法更快的渐近速度找到重叠并生成组装字符串图。
标准的重叠组装方法的时间复杂度为 O(N(2)),其中 N 是读长的总和。我们使用源自 Burrows-Wheeler 变换的 Ferragina-Manzini 索引 (FM-index) 在一组读长中找到至少 tau 长的重叠。除了一种找到所有重叠然后实现传递约简以生成字符串图的方法外,我们还展示了如何直接仅输出不可约重叠,大大缩小内存需求并将计算时间减少到 O(N),与深度无关。基于重叠的组装方法自然可以处理混合长度的读长集,包括毛细管读长或第三代测序技术承诺的长读长。我们在这里提出的算法为基于重叠的组装方法的开发铺平了道路,这些方法可以扩展到整个脊椎动物基因组从头组装。