Institut Pasteur, Unité postulante de Pathogenèse de Helicobacter, Paris, France.
BMC Genomics. 2010 Jun 10;11:368. doi: 10.1186/1471-2164-11-368.
Helicobacter pylori infection is associated with several gastro-duodenal inflammatory diseases of various levels of severity. To determine whether certain combinations of genetic markers can be used to predict the clinical source of the infection, we analyzed well documented and geographically homogenous clinical isolates using a comparative genomics approach.
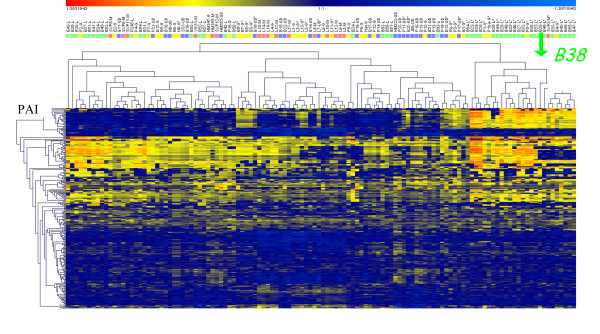
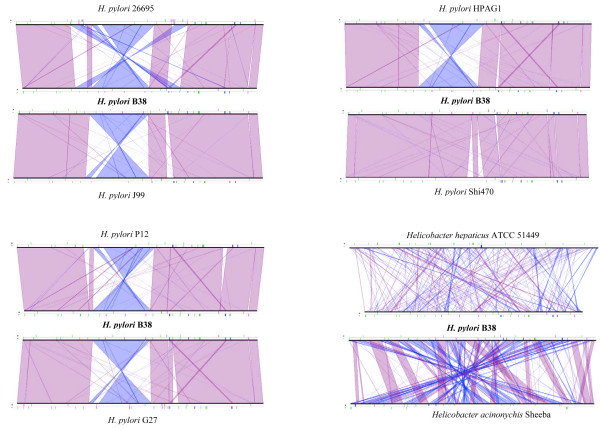
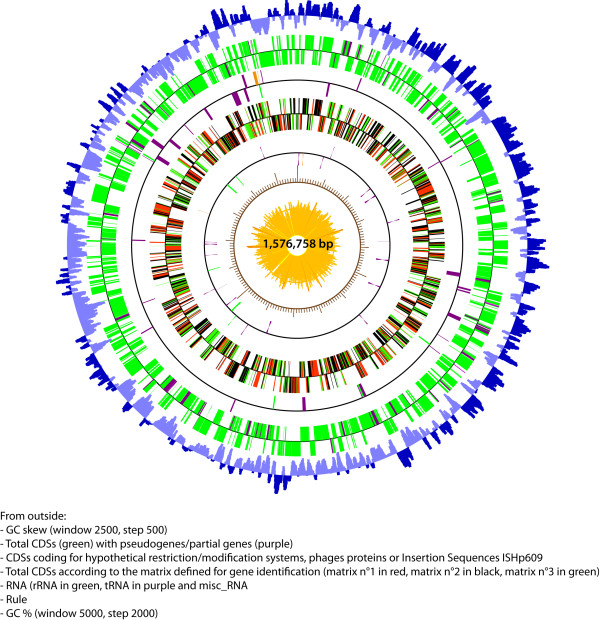
A set of 254 H. pylori genes was used to perform array-based comparative genomic hybridization among 120 French H. pylori strains associated with chronic gastritis (n = 33), duodenal ulcers (n = 27), intestinal metaplasia (n = 17) or gastric extra-nodal marginal zone B-cell MALT lymphoma (n = 43). Hierarchical cluster analyses of the DNA hybridization values allowed us to identify a homogeneous subpopulation of strains that clustered exclusively with cagPAI minus MALT lymphoma isolates. The genome sequence of B38, a representative of this MALT lymphoma strain-cluster, was completed, fully annotated, and compared with the six previously released H. pylori genomes (i.e. J99, 26695, HPAG1, P12, G27 and Shi470). B38 has the smallest H. pylori genome described thus far (1,576,758 base pairs containing 1,528 CDSs); it contains the vacAs2m2 allele and lacks the genes encoding the major virulence factors (absence of cagPAI, babB, babC, sabB, and homB). Comparative genomics led to the identification of very few sequences that are unique to the B38 strain (9 intact CDSs and 7 pseudogenes). Pair-wise genomic synteny comparisons between B38 and the 6 H. pylori sequenced genomes revealed an almost complete co-linearity, never seen before between the genomes of strain Shi470 (a Peruvian isolate) and B38.
These isolates are deprived of the main H. pylori virulence factors characterized previously, but are nonetheless associated with gastric neoplasia.
幽门螺杆菌感染与多种严重程度的胃十二指肠炎症性疾病有关。为了确定某些遗传标记的组合是否可用于预测感染的临床来源,我们使用比较基因组学方法分析了记录良好且地理位置同质的临床分离株。
使用一组 254 个幽门螺杆菌基因,在 120 株法国幽门螺杆菌菌株中进行基于阵列的比较基因组杂交,这些菌株与慢性胃炎(n = 33)、十二指肠溃疡(n = 27)、肠上皮化生(n = 17)或胃结外边缘区 B 细胞黏膜相关淋巴组织淋巴瘤(n = 43)有关。DNA 杂交值的层次聚类分析使我们能够识别出一组与 cagPAI 缺失 MALT 淋巴瘤分离株完全聚类的同型菌株亚群。B38 是该 MALT 淋巴瘤株聚类的代表,其基因组序列已完成、完全注释,并与之前发布的六个幽门螺杆菌基因组(即 J99、26695、HPAG1、P12、G27 和 Shi470)进行了比较。B38 具有迄今为止描述的最小的幽门螺杆菌基因组(1,576,758 个碱基对,包含 1,528 个 CDS);它包含 vacAs2m2 等位基因,并且缺乏主要毒力因子的基因(cagPAI、babB、babC、sabB 和 homB 缺失)。比较基因组学导致仅鉴定出 B38 株特有的极少数序列(9 个完整 CDS 和 7 个假基因)。B38 与 6 个已测序的幽门螺杆菌基因组之间的基因组成对同线性比较显示,以前从未在 Shi470 菌株(秘鲁分离株)和 B38 之间见过基因组之间几乎完全的共线性。
这些分离株缺乏以前特征的主要幽门螺杆菌毒力因子,但仍与胃肿瘤有关。