CSIRO Plant Industry, Black Mountain Laboratories, Canberra, Australia.
BMC Genomics. 2012 Sep 17;13:484. doi: 10.1186/1471-2164-13-484.
RNA sequencing (RNA-Seq) has emerged as a powerful approach for the detection of differential gene expression with both high-throughput and high resolution capabilities possible depending upon the experimental design chosen. Multiplex experimental designs are now readily available, these can be utilised to increase the numbers of samples or replicates profiled at the cost of decreased sequencing depth generated per sample. These strategies impact on the power of the approach to accurately identify differential expression. This study presents a detailed analysis of the power to detect differential expression in a range of scenarios including simulated null and differential expression distributions with varying numbers of biological or technical replicates, sequencing depths and analysis methods.
Differential and non-differential expression datasets were simulated using a combination of negative binomial and exponential distributions derived from real RNA-Seq data. These datasets were used to evaluate the performance of three commonly used differential expression analysis algorithms and to quantify the changes in power with respect to true and false positive rates when simulating variations in sequencing depth, biological replication and multiplex experimental design choices.
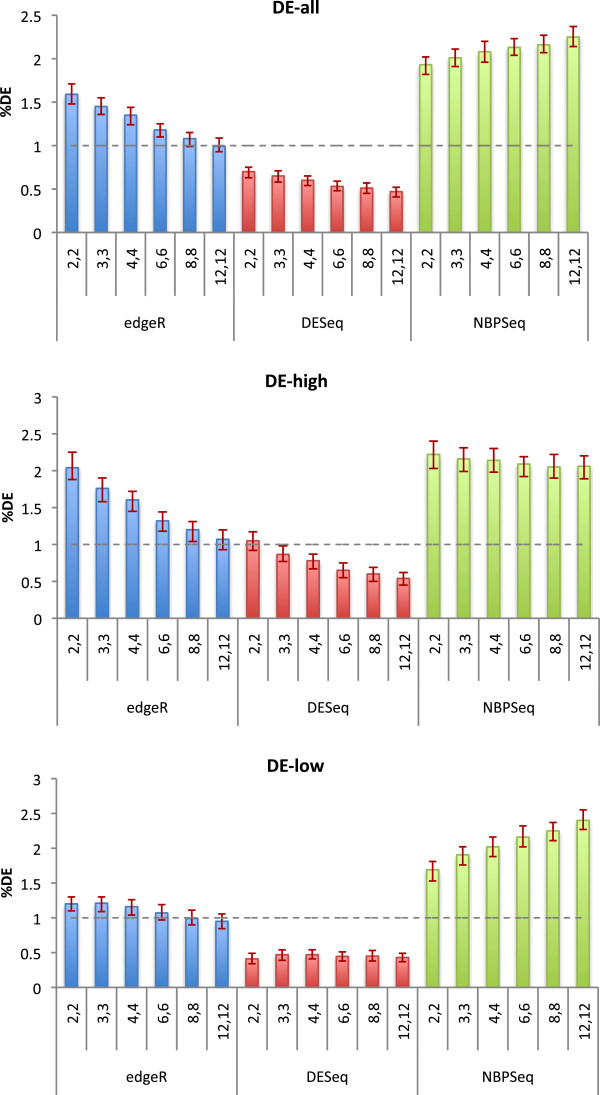
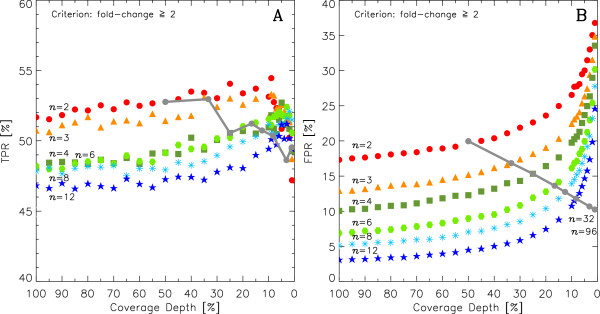
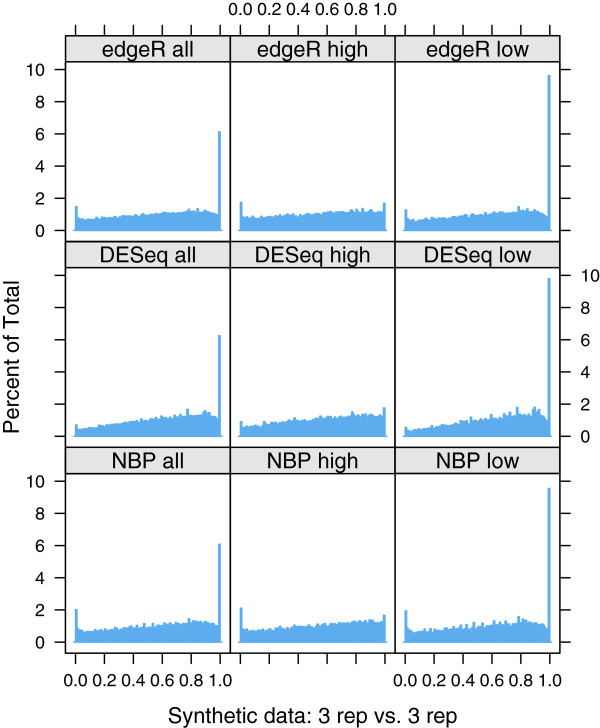
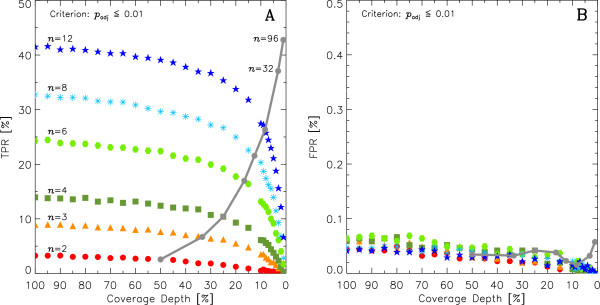
This work quantitatively explores comparisons between contemporary analysis tools and experimental design choices for the detection of differential expression using RNA-Seq. We found that the DESeq algorithm performs more conservatively than edgeR and NBPSeq. With regard to testing of various experimental designs, this work strongly suggests that greater power is gained through the use of biological replicates relative to library (technical) replicates and sequencing depth. Strikingly, sequencing depth could be reduced as low as 15% without substantial impacts on false positive or true positive rates.
RNA 测序(RNA-Seq)已经成为一种强大的方法,可以通过选择的实验设计实现高通量和高分辨率的差异基因表达检测。现在已经有了多种多重实验设计,这些设计可以用来增加样本或重复的数量,而代价是每个样本产生的测序深度降低。这些策略会影响该方法准确识别差异表达的能力。本研究详细分析了在各种情况下检测差异表达的能力,包括模拟的零和差异表达分布,以及具有不同数量的生物学或技术重复、测序深度和分析方法的情况。
使用从真实 RNA-Seq 数据中得出的负二项式和指数分布的组合来模拟差异和非差异表达数据集。这些数据集用于评估三种常用的差异表达分析算法的性能,并在模拟测序深度、生物学重复和多重实验设计选择变化时,量化与真实和假阳性率相关的功率变化。
这项工作定量地探索了使用 RNA-Seq 检测差异表达时,当代分析工具和实验设计选择之间的比较。我们发现,DESeq 算法比 edgeR 和 NBPSeq 更保守。关于各种实验设计的测试,这项工作强烈表明,通过使用生物学重复而不是文库(技术)重复和测序深度,可以获得更大的功率。引人注目的是,测序深度可以降低到 15%以下,而不会对假阳性或真阳性率产生实质性影响。