MIT Computer Science and Artificial Intelligence Laboratory, Cambridge, Massachusetts, USA.
Nat Biotechnol. 2010 Aug;28(8):817-25. doi: 10.1038/nbt.1662. Epub 2010 Jul 25.
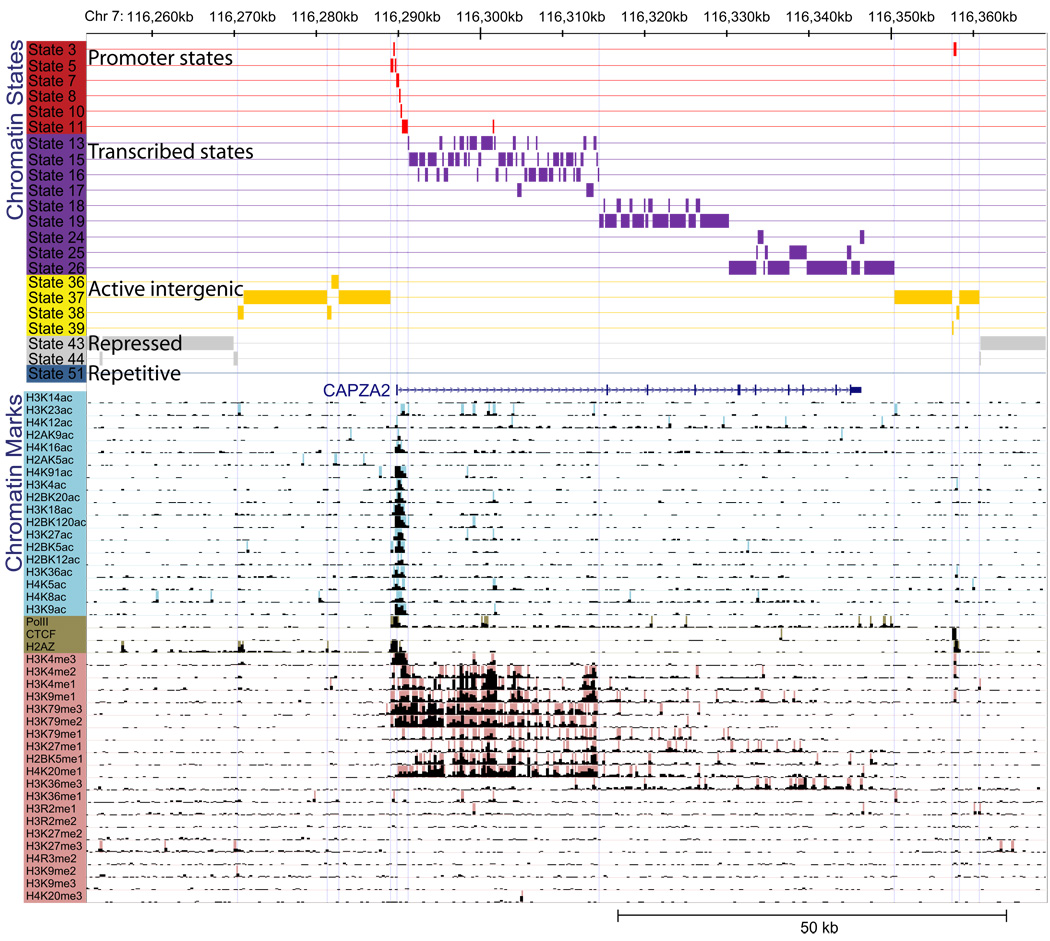
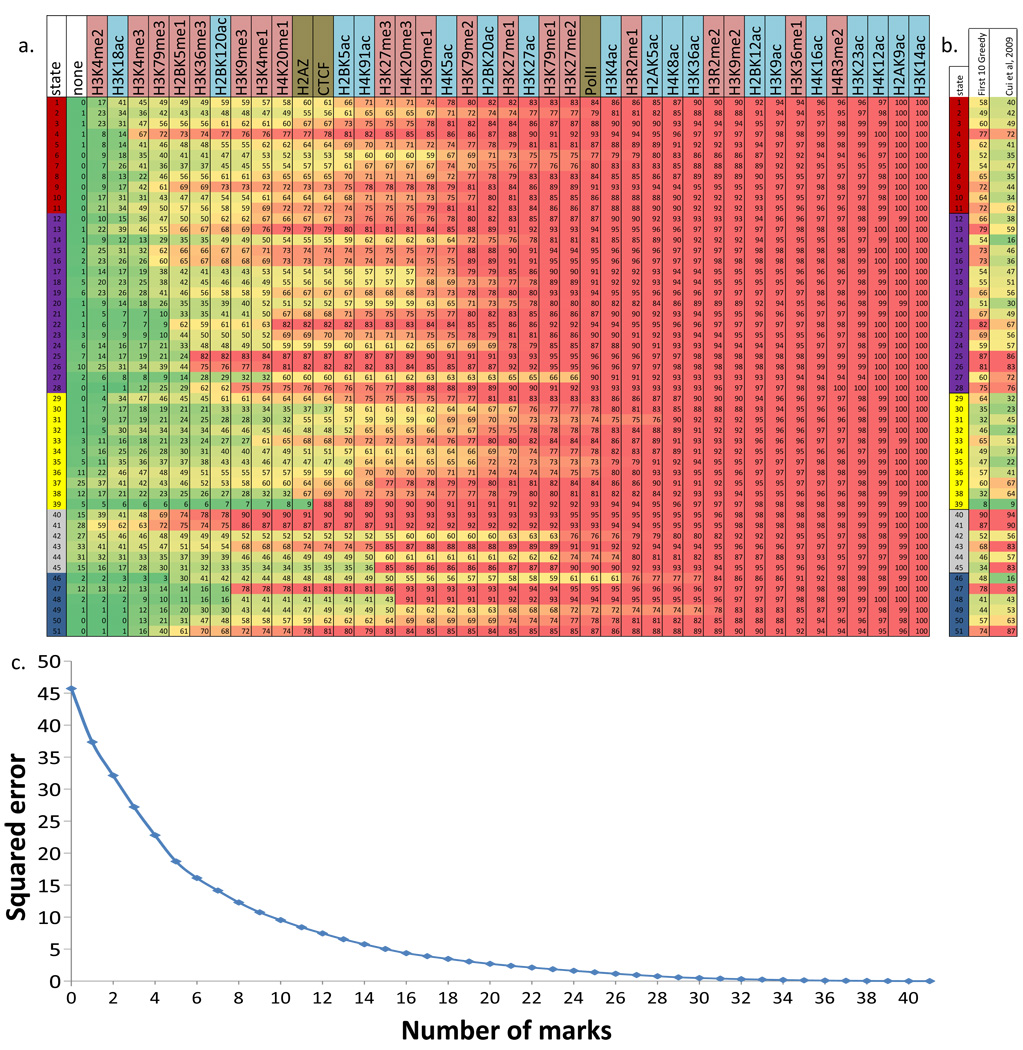
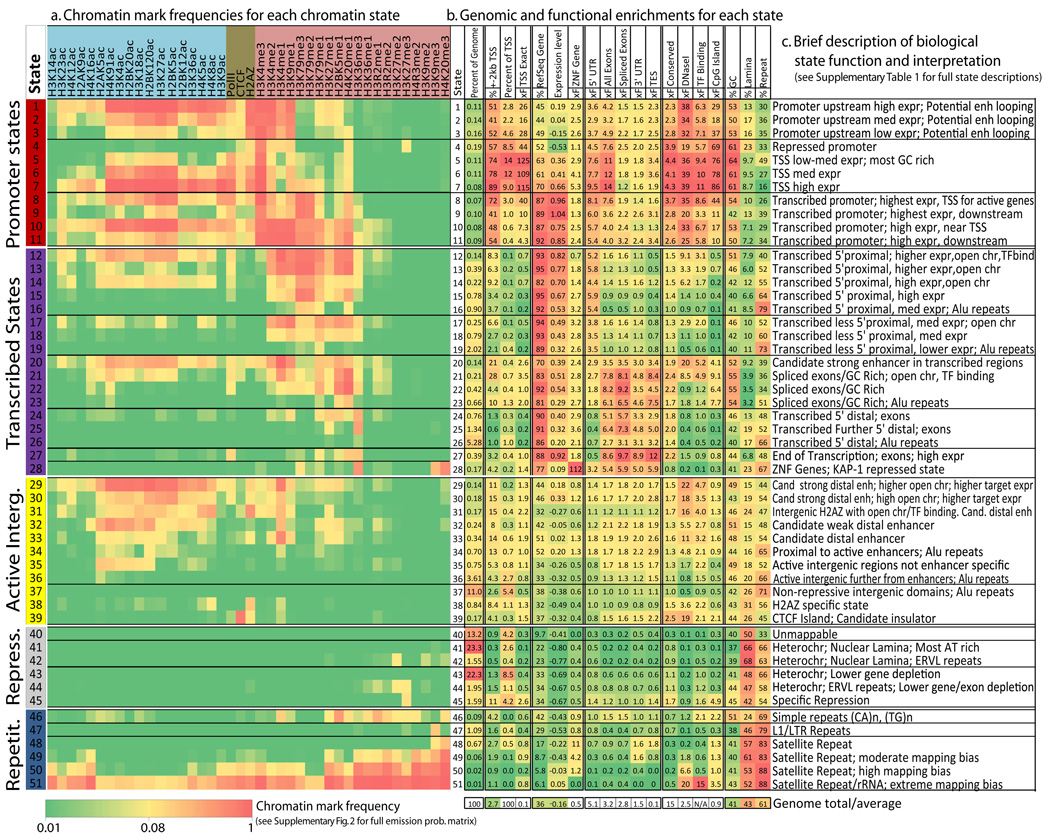
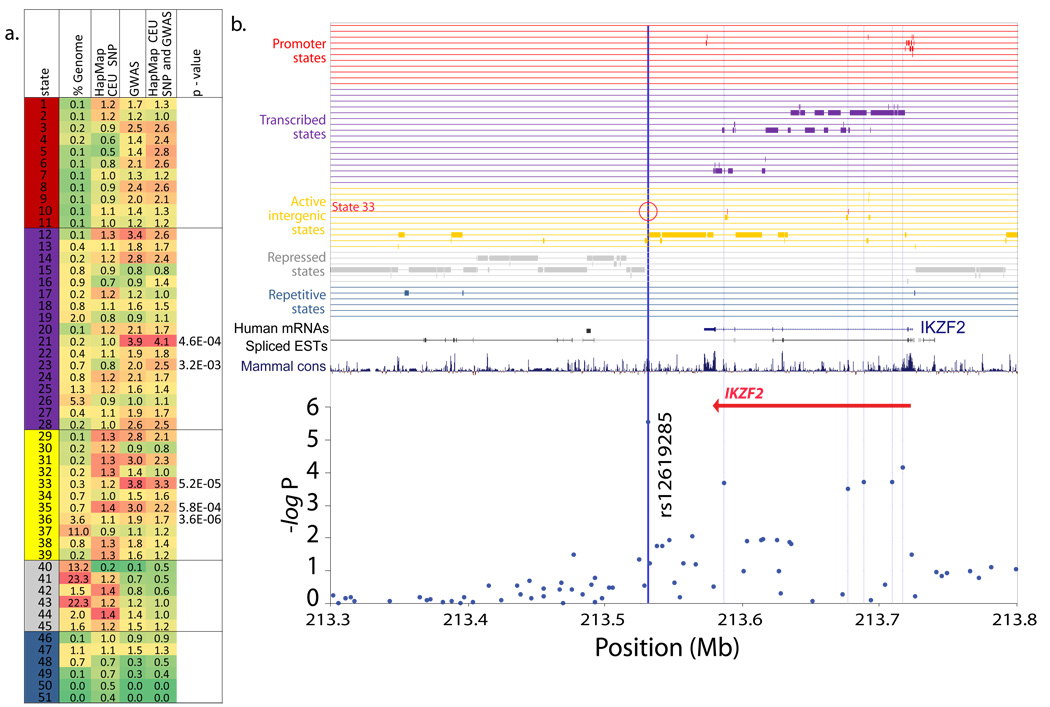
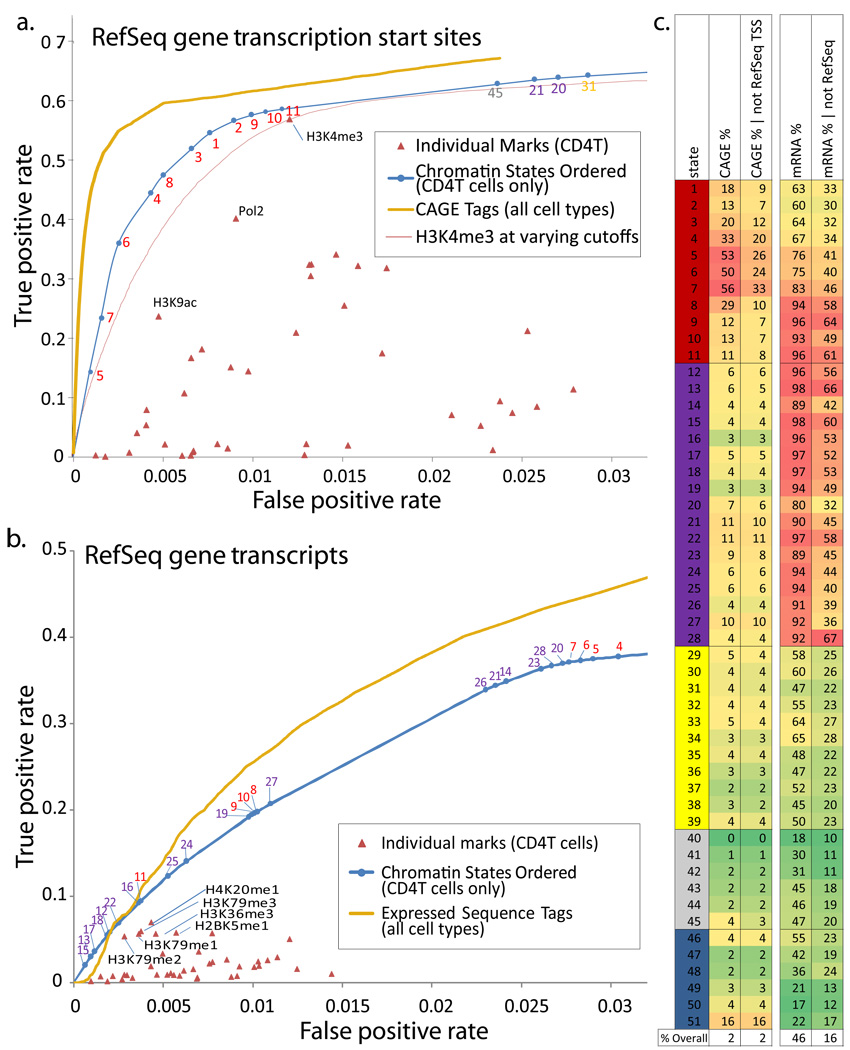
A plethora of epigenetic modifications have been described in the human genome and shown to play diverse roles in gene regulation, cellular differentiation and the onset of disease. Although individual modifications have been linked to the activity levels of various genetic functional elements, their combinatorial patterns are still unresolved and their potential for systematic de novo genome annotation remains untapped. Here, we use a multivariate Hidden Markov Model to reveal 'chromatin states' in human T cells, based on recurrent and spatially coherent combinations of chromatin marks. We define 51 distinct chromatin states, including promoter-associated, transcription-associated, active intergenic, large-scale repressed and repeat-associated states. Each chromatin state shows specific enrichments in functional annotations, sequence motifs and specific experimentally observed characteristics, suggesting distinct biological roles. This approach provides a complementary functional annotation of the human genome that reveals the genome-wide locations of diverse classes of epigenetic function.
大量的表观遗传修饰已在人类基因组中被描述,并被证明在基因调控、细胞分化和疾病发生中发挥着多样化的作用。尽管个别修饰与各种遗传功能元件的活性水平有关,但它们的组合模式仍未解决,其用于系统从头基因组注释的潜力尚未被挖掘。在这里,我们使用多元隐马尔可夫模型(multivariate Hidden Markov Model),基于染色质标记的重复和空间一致的组合,在人类 T 细胞中揭示“染色质状态”。我们定义了 51 种不同的染色质状态,包括与启动子相关、转录相关、活跃的基因间区、大规模抑制和重复相关的状态。每个染色质状态在功能注释、序列基序和特定实验观察到的特征中都有特定的富集,这表明它们具有不同的生物学作用。这种方法提供了人类基因组的互补功能注释,揭示了各种类别的表观遗传功能在全基因组上的位置。