1Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA 90095 USA.
2Department of Biological Chemistry, University of California, Los Angeles, Los Angeles, CA 90095 USA.
Commun Biol. 2019 Jul 2;2:248. doi: 10.1038/s42003-019-0488-1. eCollection 2019.
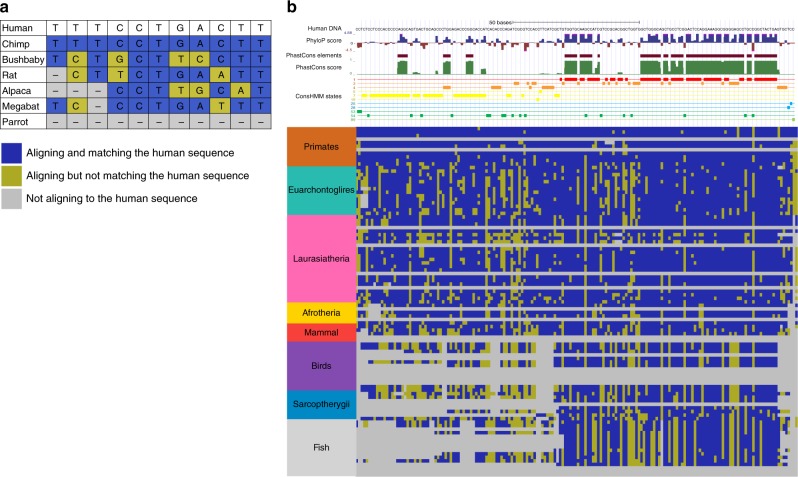
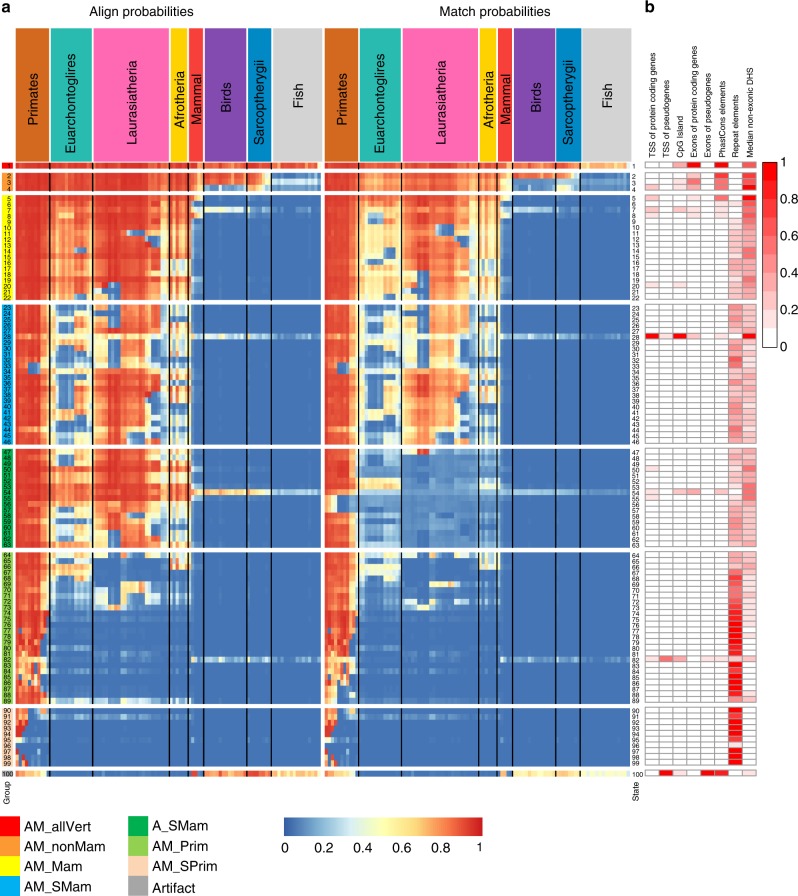
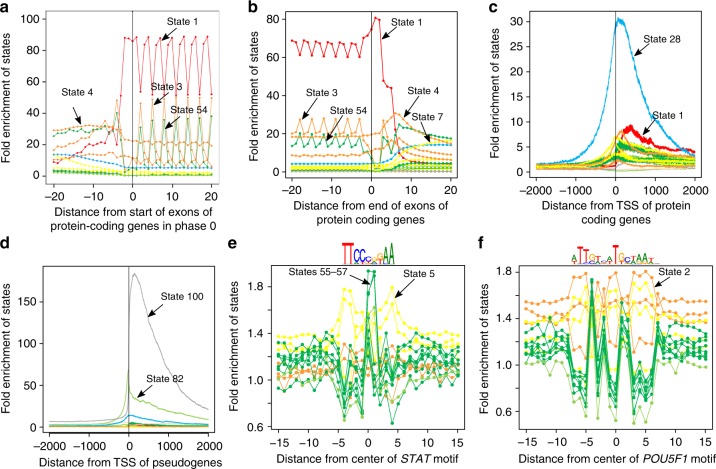
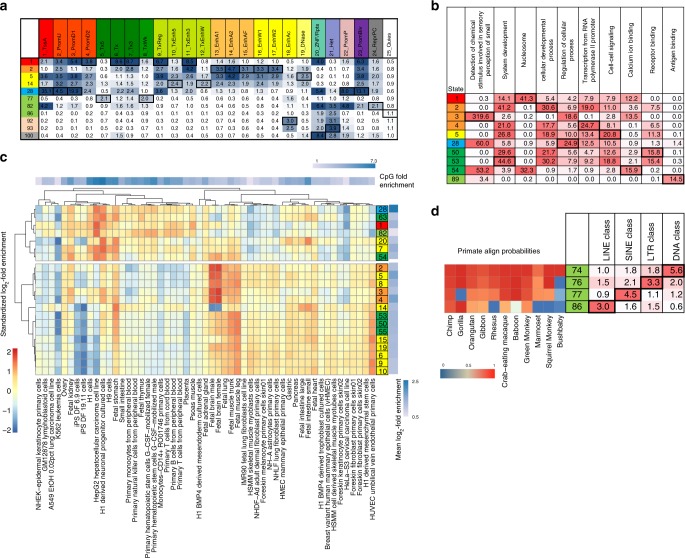
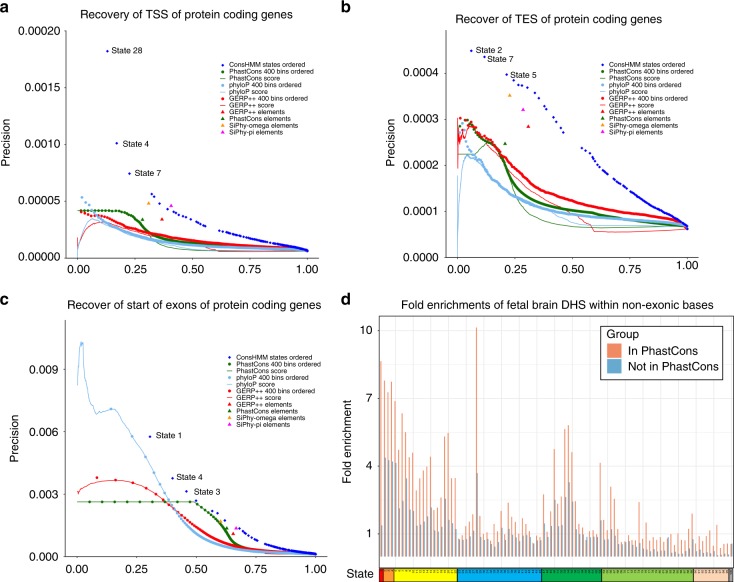
Comparative genomics sequence data is an important source of information for interpreting genomes. Genome-wide annotations based on this data have largely focused on univariate scores or binary elements of evolutionary constraint. Here we present a complementary whole genome annotation approach, ConsHMM, which applies a multivariate hidden Markov model to learn de novo 'conservation states' based on the combinatorial and spatial patterns of which species align to and match a reference genome in a multiple species DNA sequence alignment. We applied ConsHMM to a 100-way vertebrate sequence alignment to annotate the human genome at single nucleotide resolution into 100 conservation states. These states have distinct enrichments for other genomic information including gene annotations, chromatin states, repeat families, and bases prioritized by various variant prioritization scores. Constrained elements have distinct heritability partitioning enrichments depending on their conservation state assignment. ConsHMM conservation states are a resource for analyzing genomes and genetic variants.
比较基因组序列数据是解释基因组的重要信息来源。基于这些数据的全基因组注释主要集中在单变量分数或进化约束的二进制元素上。在这里,我们提出了一种互补的全基因组注释方法 ConsHMM,它应用多变量隐马尔可夫模型来学习基于组合和空间模式的新的“保守状态”,这些模式基于多物种 DNA 序列比对中物种与参考基因组的对齐和匹配。我们将 ConsHMM 应用于 100 种脊椎动物序列比对,以单核苷酸分辨率将人类基因组注释为 100 种保守状态。这些状态在其他基因组信息(包括基因注释、染色质状态、重复家族和各种变体优先级得分优先的碱基)方面有明显的富集。根据保守状态分配,受约束的元素具有不同的遗传分割富集。ConsHMM 保守状态是分析基因组和遗传变异的资源。