Ernst Jason, Kellis Manolis
Department of Biological Chemistry, University of California, Los Angeles, Los Angeles, California, USA.
Department of Computer Science, University of California, Los Angeles, Los Angeles, California, USA.
Nat Protoc. 2017 Dec;12(12):2478-2492. doi: 10.1038/nprot.2017.124. Epub 2017 Nov 9.
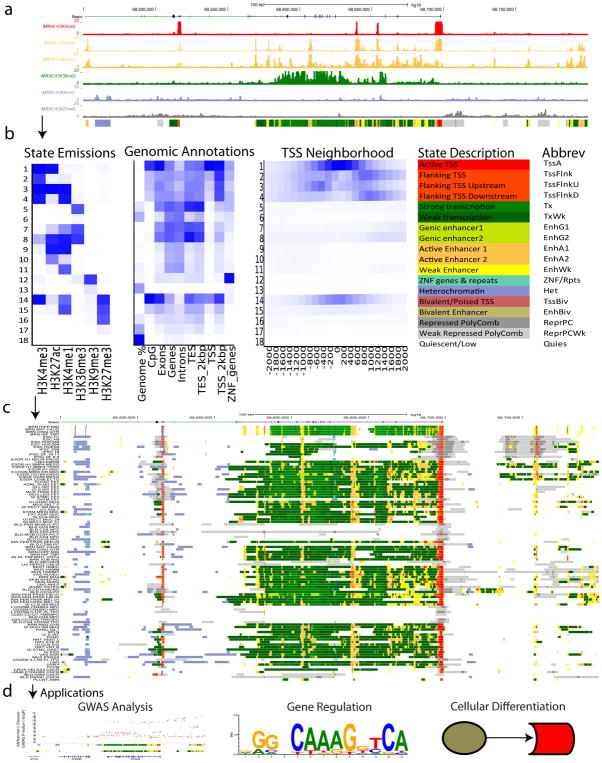
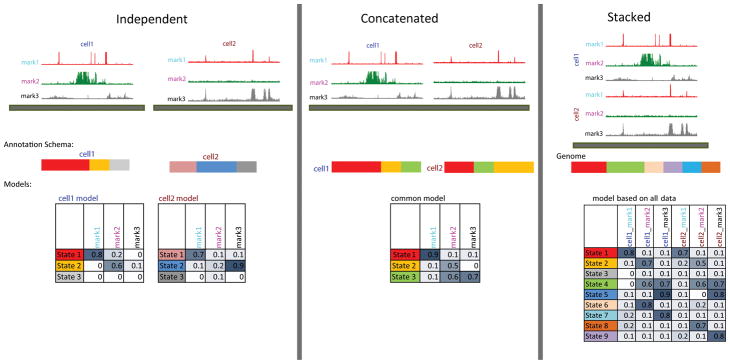
Noncoding DNA regions have central roles in human biology, evolution, and disease. ChromHMM helps to annotate the noncoding genome using epigenomic information across one or multiple cell types. It combines multiple genome-wide epigenomic maps, and uses combinatorial and spatial mark patterns to infer a complete annotation for each cell type. ChromHMM learns chromatin-state signatures using a multivariate hidden Markov model (HMM) that explicitly models the combinatorial presence or absence of each mark. ChromHMM uses these signatures to generate a genome-wide annotation for each cell type by calculating the most probable state for each genomic segment. ChromHMM provides an automated enrichment analysis of the resulting annotations to facilitate the functional interpretations of each chromatin state. ChromHMM is distinguished by its modeling emphasis on combinations of marks, its tight integration with downstream functional enrichment analyses, its speed, and its ease of use. Chromatin states are learned, annotations are produced, and enrichments are computed within 1 d.
非编码DNA区域在人类生物学、进化和疾病中起着核心作用。ChromHMM有助于利用跨一种或多种细胞类型的表观基因组信息对非编码基因组进行注释。它结合了多个全基因组表观基因组图谱,并使用组合和空间标记模式来推断每种细胞类型的完整注释。ChromHMM使用多变量隐马尔可夫模型(HMM)学习染色质状态特征,该模型明确地对每个标记的组合存在或不存在进行建模。ChromHMM通过计算每个基因组片段的最可能状态,使用这些特征为每种细胞类型生成全基因组注释。ChromHMM对所得注释进行自动富集分析,以促进对每种染色质状态的功能解释。ChromHMM的特点在于其对标记组合的建模重点、与下游功能富集分析的紧密整合、速度以及易用性。染色质状态得以学习,注释得以生成,富集分析在1天内即可完成。