Key Laboratory of Systems Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, People's Republic of China.
PLoS One. 2010 Jul 30;5(7):e11900. doi: 10.1371/journal.pone.0011900.
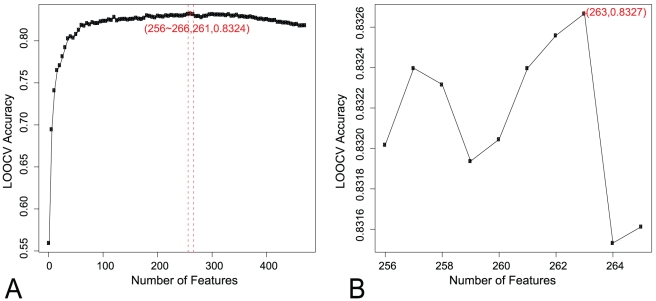
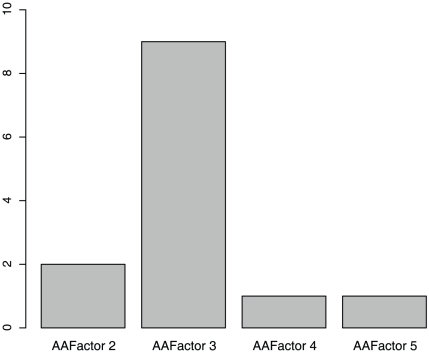
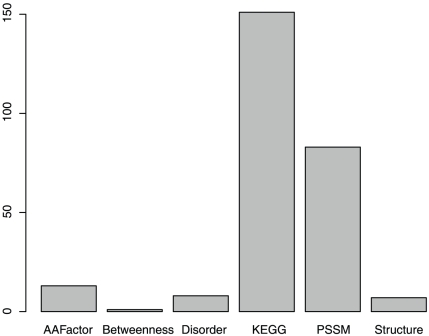
Non-synonymous SNPs (nsSNPs), also known as Single Amino acid Polymorphisms (SAPs) account for the majority of human inherited diseases. It is important to distinguish the deleterious SAPs from neutral ones. Most traditional computational methods to classify SAPs are based on sequential or structural features. However, these features cannot fully explain the association between a SAP and the observed pathophysiological phenotype. We believe the better rationale for deleterious SAP prediction should be: If a SAP lies in the protein with important functions and it can change the protein sequence and structure severely, it is more likely related to disease. So we established a method to predict deleterious SAPs based on both protein interaction network and traditional hybrid properties. Each SAP is represented by 472 features that include sequential features, structural features and network features. Maximum Relevance Minimum Redundancy (mRMR) method and Incremental Feature Selection (IFS) were applied to obtain the optimal feature set and the prediction model was Nearest Neighbor Algorithm (NNA). In jackknife cross-validation, 83.27% of SAPs were correctly predicted when the optimized 263 features were used. The optimized predictor with 263 features was also tested in an independent dataset and the accuracy was still 80.00%. In contrast, SIFT, a widely used predictor of deleterious SAPs based on sequential features, has a prediction accuracy of 71.05% on the same dataset. In our study, network features were found to be most important for accurate prediction and can significantly improve the prediction performance. Our results suggest that the protein interaction context could provide important clues to help better illustrate SAP's functional association. This research will facilitate the post genome-wide association studies.
非同义 SNP(nsSNP),又称单氨基酸多态性(SAP),占人类遗传性疾病的绝大多数。区分有害 SAP 与中性 SAP 非常重要。大多数传统的 SAP 分类计算方法都是基于序列或结构特征。然而,这些特征并不能完全解释 SAP 与观察到的病理生理表型之间的关联。我们认为,有害 SAP 预测的更好原理应该是:如果 SAP 位于具有重要功能的蛋白质中,并且能够严重改变蛋白质序列和结构,那么它与疾病的相关性就更高。因此,我们建立了一种基于蛋白质相互作用网络和传统混合特性的有害 SAP 预测方法。每个 SAP 由 472 个特征表示,包括序列特征、结构特征和网络特征。采用最大相关最小冗余(mRMR)方法和增量特征选择(IFS)来获得最佳特征集,并采用最近邻算法(NNA)构建预测模型。在 Jackknife 交叉验证中,当使用优化的 263 个特征时,83.27%的 SAP 被正确预测。优化后的 263 个特征的预测器也在独立数据集上进行了测试,准确率仍然为 80.00%。相比之下,基于序列特征的广泛使用的有害 SAP 预测器 SIFT 在同一数据集上的预测准确率为 71.05%。在我们的研究中,网络特征被发现对准确预测最重要,并且可以显著提高预测性能。我们的结果表明,蛋白质相互作用背景可以提供重要线索,有助于更好地说明 SAP 的功能关联。这项研究将促进全基因组关联研究之后的工作。