Hu Jing, Yan Changhui
Department of Computer Science, Utah State University, Logan, UT 84322, USA.
BMC Bioinformatics. 2008 Jun 27;9:297. doi: 10.1186/1471-2105-9-297.
As the number of non-synonymous single nucleotide polymorphisms (nsSNPs), also known as single amino acid polymorphisms (SAPs), increases rapidly, computational methods that can distinguish disease-causing SAPs from neutral SAPs are needed. Many methods have been developed to distinguish disease-causing SAPs based on both structural and sequence features of the mutation point. One limitation of these methods is that they are not applicable to the cases where protein structures are not available. In this study, we explore the feasibility of classifying SAPs into disease-causing and neutral mutations using only information derived from protein sequence.
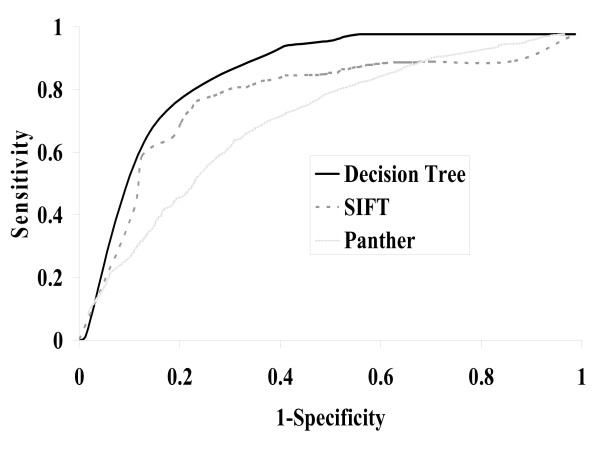
We compiled a set of 686 features that were derived from protein sequence. For each feature, the distance between the wild-type residue and mutant-type residue was computed. Then a greedy approach was used to select the features that were useful for the classification of SAPs. 10 features were selected. Using the selected features, a decision tree method can achieve 82.6% overall accuracy with 0.607 Matthews Correlation Coefficient (MCC) in cross-validation. When tested on an independent set that was not seen by the method during the training and feature selection, the decision tree method achieves 82.6% overall accuracy with 0.604 MCC. We also evaluated the proposed method on all SAPs obtained from the Swiss-Prot, the method achieves 0.42 MCC with 73.2% overall accuracy. This method allows users to make reliable predictions when protein structures are not available. Different from previous studies, in which only a small set of features were arbitrarily chosen and considered, here we used an automated method to systematically discover useful features from a large set of features well-annotated in public databases.
The proposed method is a useful tool for the classification of SAPs, especially, when the structure of the protein is not available.
随着非同义单核苷酸多态性(nsSNPs),也称为单氨基酸多态性(SAPs)的数量迅速增加,需要能够区分致病SAPs和中性SAPs的计算方法。已经开发了许多方法来基于突变点的结构和序列特征区分致病SAPs。这些方法的一个局限性是它们不适用于蛋白质结构不可用的情况。在本研究中,我们探索仅使用从蛋白质序列衍生的信息将SAPs分类为致病突变和中性突变的可行性。
我们汇编了一组从蛋白质序列衍生的686个特征。对于每个特征,计算野生型残基和突变型残基之间的距离。然后使用贪婪方法选择对SAPs分类有用的特征。选择了10个特征。使用所选特征,决策树方法在交叉验证中可以达到82.6%的总体准确率和0.607的马修斯相关系数(MCC)。在训练和特征选择期间该方法未见过的独立集上进行测试时,决策树方法达到82.6%的总体准确率和0.604的MCC。我们还在从Swiss-Prot获得的所有SAPs上评估了所提出的方法,该方法达到0.42的MCC和73.2%的总体准确率。当蛋白质结构不可用时,该方法允许用户进行可靠的预测。与以前的研究不同,在以前的研究中仅任意选择和考虑了一小部分特征,在这里我们使用一种自动化方法从公共数据库中注释良好的大量特征中系统地发现有用的特征。
所提出的方法是用于SAPs分类的有用工具,特别是当蛋白质结构不可用时。