Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, PR China.
BMC Bioinformatics. 2011 Jan 12;12:14. doi: 10.1186/1471-2105-12-14.
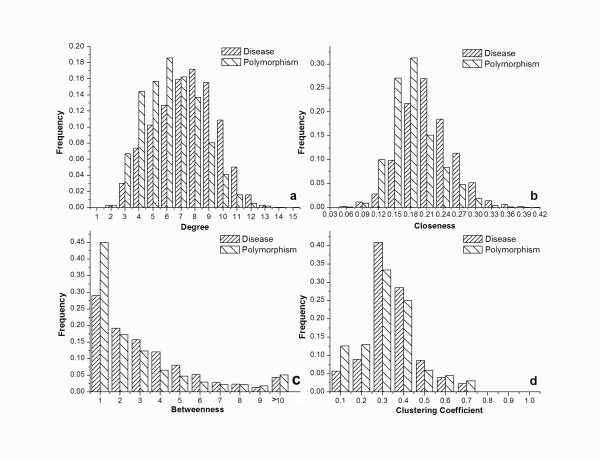
The rapid accumulation of data on non-synonymous single nucleotide polymorphisms (nsSNPs, also called SAPs) should allow us to further our understanding of the underlying disease-associated mechanisms. Here, we use complex networks to study the role of an amino acid in both local and global structures and determine the extent to which disease-associated and polymorphic SAPs differ in terms of their interactions to other residues.
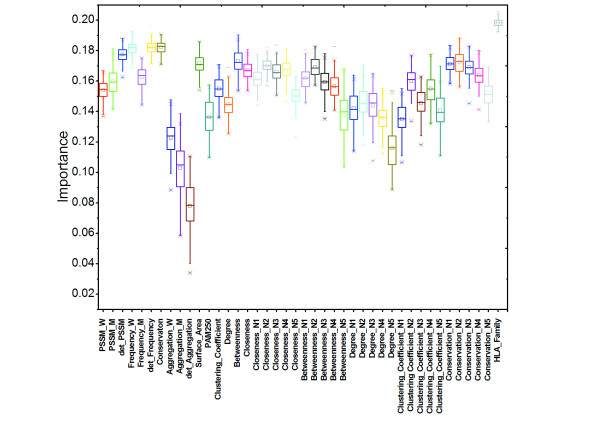
We found that SAPs can be well characterized by network topological features. Mutations are probably disease-associated when they occur at a site with a high centrality value and/or high degree value in a protein structure network. We also discovered that study of the neighboring residues around a mutation site can help to determine whether the mutation is disease-related or not. We compiled a dataset from the Swiss-Prot variant pages and constructed a model to predict disease-associated SAPs based on the random forest algorithm. The values of total accuracy and MCC were 83.0% and 0.64, respectively, as determined by 5-fold cross-validation. With an independent dataset, our model achieved a total accuracy of 80.8% and MCC of 0.59, respectively.
The satisfactory performance suggests that network topological features can be used as quantification measures to determine the importance of a site on a protein, and this approach can complement existing methods for prediction of disease-associated SAPs. Moreover, the use of this method in SAP studies would help to determine the underlying linkage between SAPs and diseases through extensive investigation of mutual interactions between residues.
非 synonymous单核苷酸多态性(nsSNP,也称为 SAP)的数据迅速积累,应该使我们能够进一步了解潜在的疾病相关机制。在这里,我们使用复杂网络来研究氨基酸在局部和全局结构中的作用,并确定与其他残基相互作用的疾病相关和多态性 SAP 之间的差异程度。
我们发现 SAP 可以很好地通过网络拓扑特征来描述。当突变发生在蛋白质结构网络中具有高中心度值和/或高度数值的位点时,突变很可能与疾病相关。我们还发现,研究突变位点周围的相邻残基有助于确定突变是否与疾病相关。我们从 Swiss-Prot 变体页面中编制了一个数据集,并基于随机森林算法构建了一个预测疾病相关 SAP 的模型。通过 5 倍交叉验证,总准确性和 MCC 的值分别为 83.0%和 0.64。使用独立数据集,我们的模型的总准确性为 80.8%,MCC 为 0.59。
令人满意的性能表明,网络拓扑特征可以用作量化措施来确定蛋白质上一个位点的重要性,并且这种方法可以补充现有的疾病相关 SAP 预测方法。此外,通过广泛研究残基之间的相互作用,在 SAP 研究中使用此方法将有助于确定 SAP 与疾病之间的潜在联系。