Department of Epidemiology, Michigan State University, East Lansing, Michigan 48824, USA.
BMC Genet. 2010 Aug 27;11:79. doi: 10.1186/1471-2156-11-79.
The genetic etiology of complex diseases in human has been commonly viewed as a complex process involving both genetic and environmental factors functioning in a complicated manner. Quite often the interactions among genetic variants play major roles in determining the susceptibility of an individual to a particular disease. Statistical methods for modeling interactions underlying complex diseases between single genetic variants (e.g. single nucleotide polymorphisms or SNPs) have been extensively studied. Recently, haplotype-based analysis has gained its popularity among genetic association studies. When multiple sequence or haplotype interactions are involved in determining an individual's susceptibility to a disease, it presents daunting challenges in statistical modeling and testing of the interaction effects, largely due to the complicated higher order epistatic complexity.
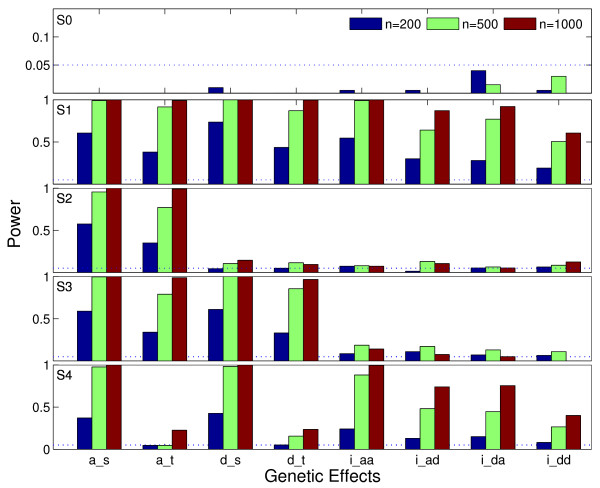
In this article, we propose a new strategy in modeling haplotype-haplotype interactions under the penalized logistic regression framework with adaptive L1-penalty. We consider interactions of sequence variants between haplotype blocks. The adaptive L1-penalty allows simultaneous effect estimation and variable selection in a single model. We propose a new parameter estimation method which estimates and selects parameters by the modified Gauss-Seidel method nested within the EM algorithm. Simulation studies show that it has low false positive rate and reasonable power in detecting haplotype interactions. The method is applied to test haplotype interactions involved in mother and offspring genome in a small for gestational age (SGA) neonates data set, and significant interactions between different genomes are detected.
As demonstrated by the simulation studies and real data analysis, the approach developed provides an efficient tool for the modeling and testing of haplotype interactions. The implementation of the method in R codes can be freely downloaded from http://www.stt.msu.edu/~cui/software.html.
人类复杂疾病的遗传病因通常被认为是一个复杂的过程,涉及遗传和环境因素以复杂的方式共同作用。遗传变异之间的相互作用通常在决定个体对特定疾病的易感性方面起着重要作用。用于建模单遗传变异(例如单核苷酸多态性或 SNPs)下复杂疾病相互作用的统计方法已经得到了广泛的研究。最近,基于单体型的分析在遗传关联研究中变得越来越流行。当多个序列或单体型相互作用参与决定个体对疾病的易感性时,由于复杂的高阶上位性复杂性,在统计建模和相互作用效应的检验方面带来了巨大的挑战。
在本文中,我们提出了一种在惩罚逻辑回归框架下基于自适应 L1 惩罚的建模单体型-单体型相互作用的新策略。我们考虑单体型块之间序列变异的相互作用。自适应 L1 惩罚允许在单个模型中同时进行效应估计和变量选择。我们提出了一种新的参数估计方法,该方法通过在 EM 算法内嵌套的修正高斯-赛德尔方法来估计和选择参数。模拟研究表明,该方法在检测单体型相互作用时具有较低的假阳性率和合理的功效。该方法应用于测试小胎龄儿(SGA)新生儿数据集母本和子代基因组中涉及的单体型相互作用,并检测到不同基因组之间的显著相互作用。
模拟研究和真实数据分析表明,所开发的方法为单体型相互作用的建模和检验提供了有效的工具。该方法的 R 代码实现可在 http://www.stt.msu.edu/~cui/software.html 上免费下载。