Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, Canada.
BMC Genomics. 2010 Sep 24;11:516. doi: 10.1186/1471-2164-11-516.
Accurate identification of synteny blocks is an important step in comparative genomics towards the understanding of genome architecture and expression. Most computer programs developed in the last decade for identifying synteny blocks have limitations. To address these limitations, we recently developed a robust program called OrthoCluster, and an online database OrthoClusterDB. In this work, we have demonstrated the application of OrthoCluster in identifying synteny blocks between the genomes of Caenorhabditis elegans and Caenorhabditis briggsae, two closely related hermaphrodite nematodes.
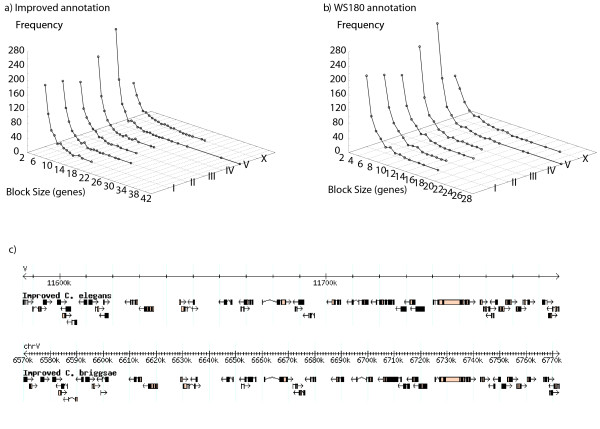
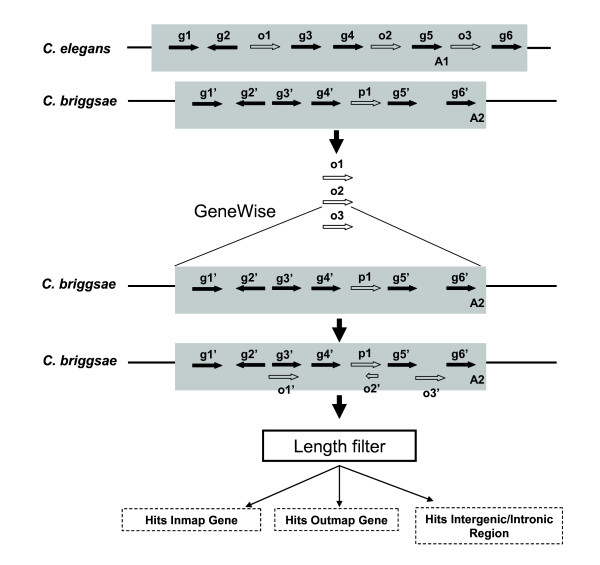
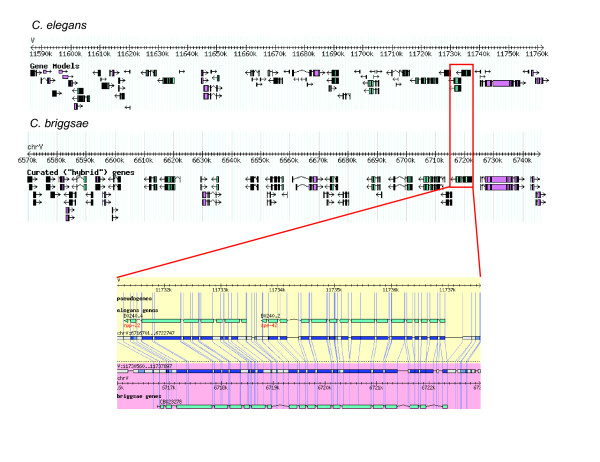
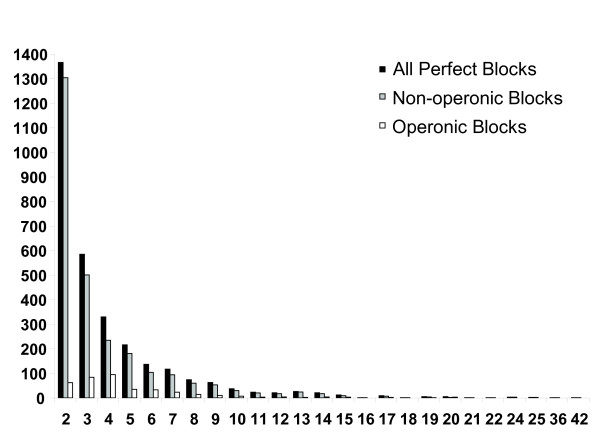
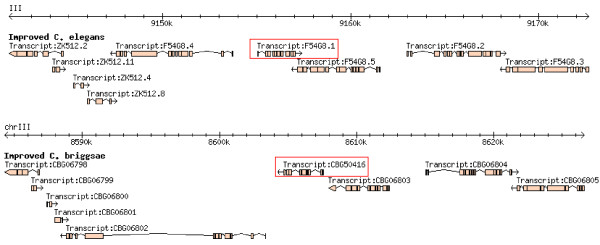
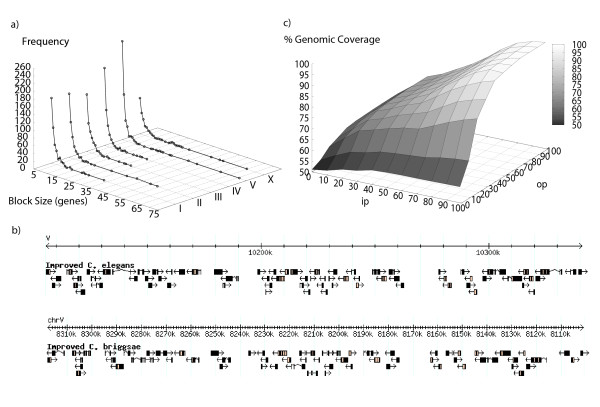
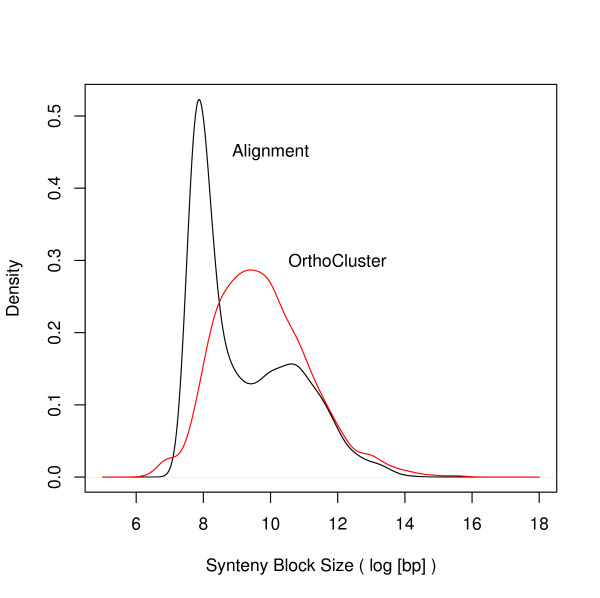
Initial identification and analysis of synteny blocks using OrthoCluster enabled us to systematically improve the genome annotation of C. elegans and C. briggsae, identifying 52 potential novel genes in C. elegans, 582 in C. briggsae, and 949 novel orthologous relationships between these two species. Using the improved annotation, we have detected 3,058 perfect synteny blocks that contain no mismatches between C. elegans and C. briggsae. Among these synteny blocks, the majority are mapped to homologous chromosomes, as previously reported. The largest perfect synteny block contains 42 genes, which spans 201.2 kb in Chromosome V of C. elegans. On average, perfect synteny blocks span 18.8 kb in length. When some mismatches (interruptions) are allowed, synteny blocks ("imperfect synteny blocks") that are much larger in size are identified. We have shown that the majority (80%) of the C. elegans and C. briggsae genomes are covered by imperfect synteny blocks. The largest imperfect synteny block spans 6.14 Mb in Chromosome X of C. elegans and there are 11 synteny blocks that are larger than 1 Mb in size. On average, imperfect synteny blocks span 63.6 kb in length, larger than previously reported.
We have demonstrated that OrthoCluster can be used to accurately identify synteny blocks and have found that synteny blocks between C. elegans and C. briggsae are almost three-folds larger than previously identified.
准确识别同线性块是比较基因组学理解基因组结构和表达的重要步骤。过去十年开发的大多数用于识别同线性块的计算机程序都存在局限性。为了解决这些限制,我们最近开发了一个名为 OrthoCluster 的强大程序和一个在线数据库 OrthoClusterDB。在这项工作中,我们展示了 OrthoCluster 在识别秀丽隐杆线虫和卡尼菲青霉这两个密切相关的雌雄同体线虫之间基因组同线性块中的应用。
使用 OrthoCluster 进行同线性块的初步识别和分析使我们能够系统地改进秀丽隐杆线虫和卡尼菲青霉的基因组注释,在秀丽隐杆线虫中鉴定出 52 个潜在的新基因,在卡尼菲青霉中鉴定出 582 个,在这两个物种之间鉴定出 949 个新的直系同源关系。使用改进的注释,我们检测到 3058 个没有秀丽隐杆线虫和卡尼菲青霉之间错配的完美同线性块。在这些同线性块中,大多数被映射到同源染色体上,如先前报道的那样。最大的完美同线性块包含 42 个基因,跨越秀丽隐杆线虫第 V 号染色体的 201.2kb。平均而言,完美同线性块的长度为 18.8kb。当允许存在一些错配(中断)时,可以识别出更大的同线性块(“不完美同线性块”)。我们已经表明,大多数(80%)秀丽隐杆线虫和卡尼菲青霉的基因组都被不完美同线性块覆盖。最大的不完美同线性块跨越秀丽隐杆线虫第 X 号染色体的 6.14Mb,有 11 个同线性块的大小大于 1Mb。平均而言,不完美同线性块的长度为 63.6kb,大于之前的报道。
我们已经证明 OrthoCluster 可以用于准确识别同线性块,并发现秀丽隐杆线虫和卡尼菲青霉之间的同线性块几乎是之前发现的三倍大。