Tang Haibao, Bomhoff Matthew D, Briones Evan, Zhang Liangsheng, Schnable James C, Lyons Eric
Center for Genomics and Biotechnology, Fujian Agriculture and Forestry University, Fuzhou, Fujian Province, China School of Plant Sciences, iPlant Collaborative, University of Arizona.
School of Plant Sciences, iPlant Collaborative, University of Arizona.
Genome Biol Evol. 2015 Nov 11;7(12):3286-98. doi: 10.1093/gbe/evv219.
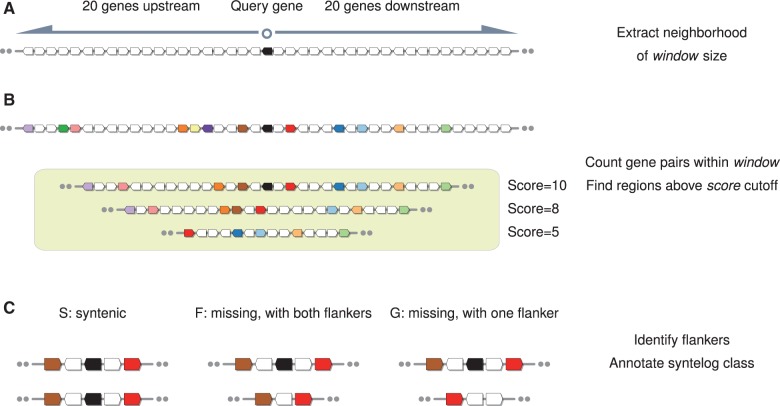
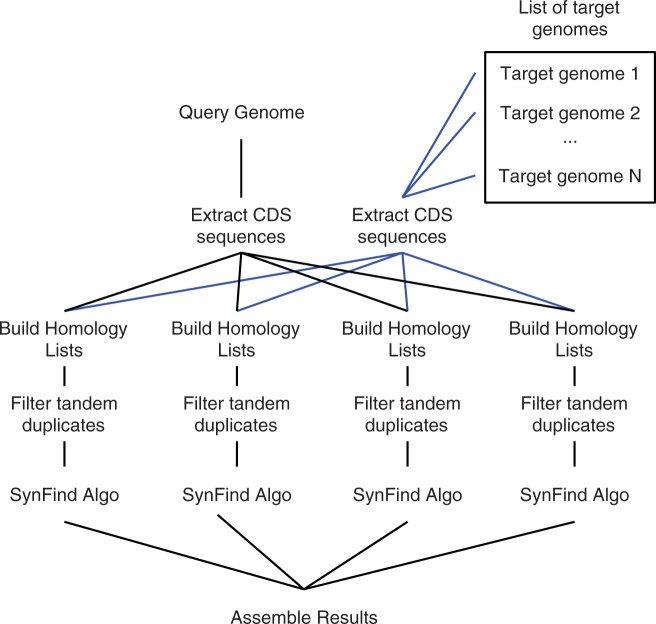
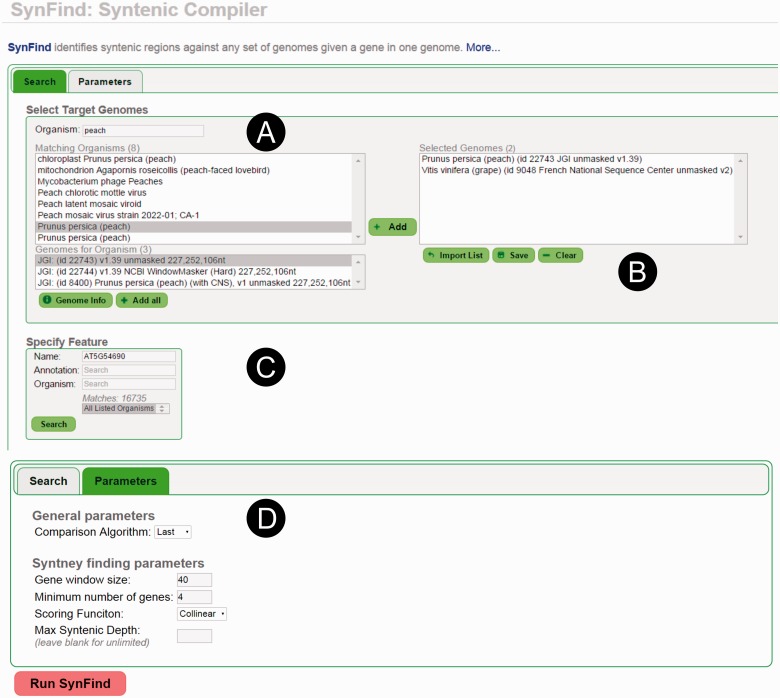
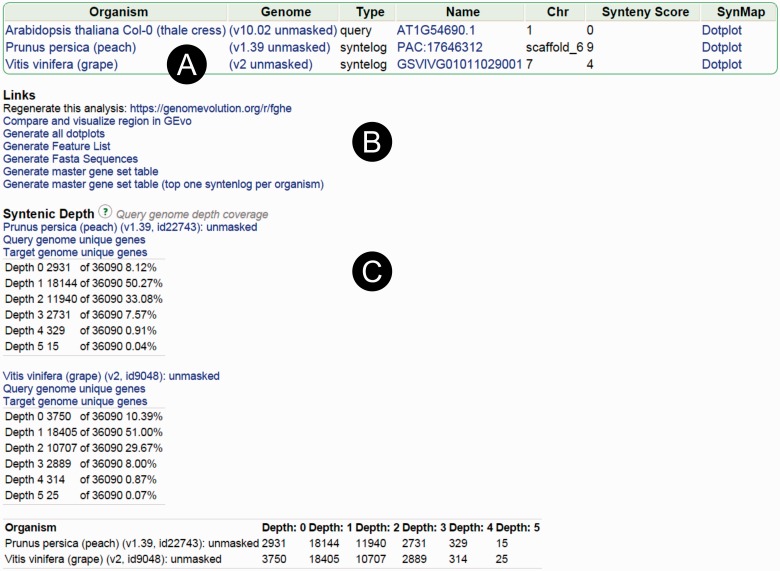
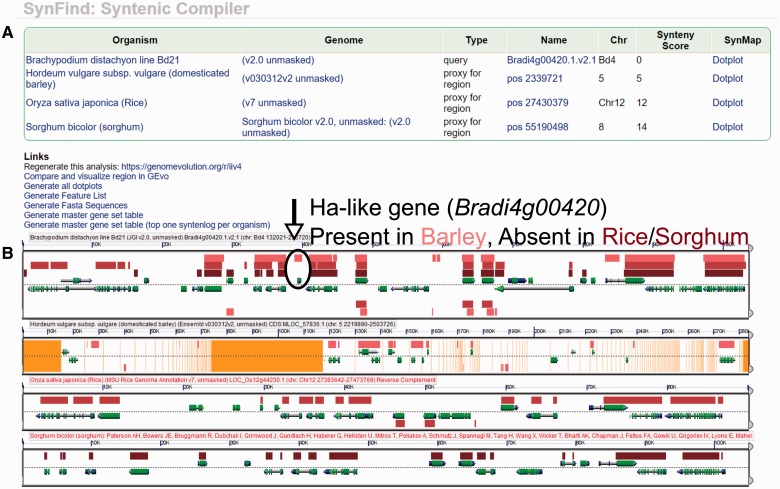
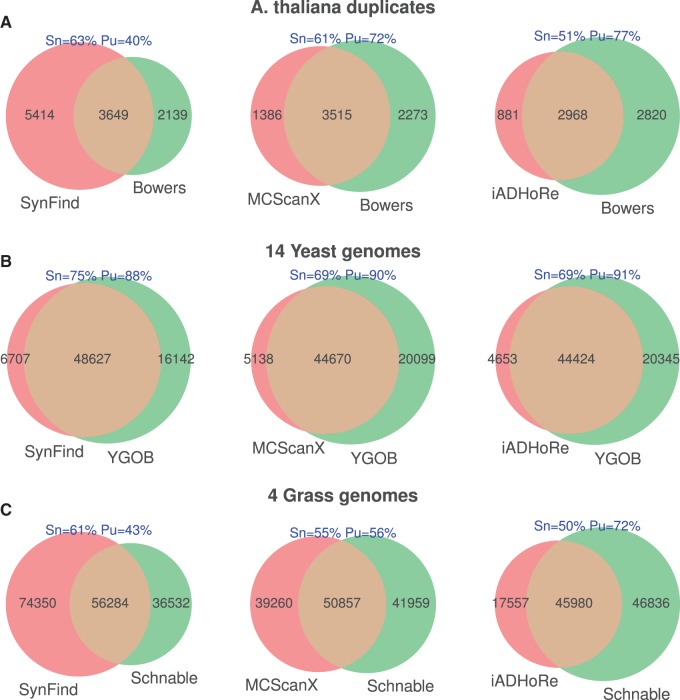
The identification of conserved syntenic regions enables discovery of predicted locations for orthologous and homeologous genes, even when no such gene is present. This capability means that synteny-based methods are far more effective than sequence similarity-based methods in identifying true-negatives, a necessity for studying gene loss and gene transposition. However, the identification of syntenic regions requires complex analyses which must be repeated for pairwise comparisons between any two species. Therefore, as the number of published genomes increases, there is a growing demand for scalable, simple-to-use applications to perform comparative genomic analyses that cater to both gene family studies and genome-scale studies. We implemented SynFind, a web-based tool that addresses this need. Given one query genome, SynFind is capable of identifying conserved syntenic regions in any set of target genomes. SynFind is capable of reporting per-gene information, useful for researchers studying specific gene families, as well as genome-wide data sets of syntenic gene and predicted gene locations, critical for researchers focused on large-scale genomic analyses. Inference of syntenic homologs provides the basis for correlation of functional changes around genes of interests between related organisms. Deployed on the CoGe online platform, SynFind is connected to the genomic data from over 15,000 organisms from all domains of life as well as supporting multiple releases of the same organism. SynFind makes use of a powerful job execution framework that promises scalability and reproducibility. SynFind can be accessed at http://genomevolution.org/CoGe/SynFind.pl. A video tutorial of SynFind using Phytophthrora as an example is available at http://www.youtube.com/watch?v=2Agczny9Nyc.
保守同线区域的识别能够发现直系同源基因和同源基因的预测位置,即便不存在此类基因。这一能力意味着,在识别真阴性方面,基于同线性的方法比基于序列相似性的方法有效得多,而这对于研究基因丢失和基因转座来说是必要的。然而,同线区域的识别需要复杂的分析,且必须针对任意两个物种之间的成对比较重复进行。因此,随着已发表基因组数量的增加,对可扩展、易于使用的应用程序的需求也在不断增长,以进行适用于基因家族研究和基因组规模研究的比较基因组分析。我们开发了SynFind,这是一个基于网络的工具,可满足这一需求。给定一个查询基因组,SynFind能够识别任何一组目标基因组中的保守同线区域。SynFind能够报告每个基因的信息,这对研究特定基因家族的研究人员很有用,同时还能提供同线基因和预测基因位置的全基因组数据集,这对专注于大规模基因组分析的研究人员至关重要。同线同源物的推断为相关生物体中感兴趣基因周围功能变化的关联提供了基础。SynFind部署在CoGe在线平台上,与来自生命所有领域的15000多种生物体的基因组数据相连,并支持同一生物体的多个版本。SynFind利用了一个强大的作业执行框架,保证了可扩展性和可重复性。可通过http://genomevolution.org/CoGe/SynFind.pl访问SynFind。以疫霉为例的SynFind视频教程可在http://www.youtube.com/watch?v=2Agczny9Nyc上获取。