Institute of Clinical Medicine, National Taiwan University, Taipei, Taiwan.
BMC Evol Biol. 2010 Sep 25;10:294. doi: 10.1186/1471-2148-10-294.
Enterovirus (EV) 71 is one of the common causative agents for hand, foot, and, mouth disease (HFMD). In recent years, the virus caused several outbreaks with high numbers of deaths and severe neurological complications. Despite the importance of these epidemics, several aspects of the evolutionary and epidemiological dynamics, including viral nucleotide variations within and between different outbreaks, rates of change in immune-related structural regions vs. non-structural regions, and forces driving the evolution of EV71, are still not clear.
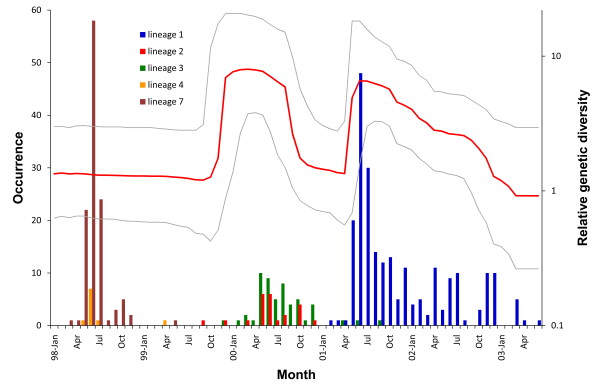
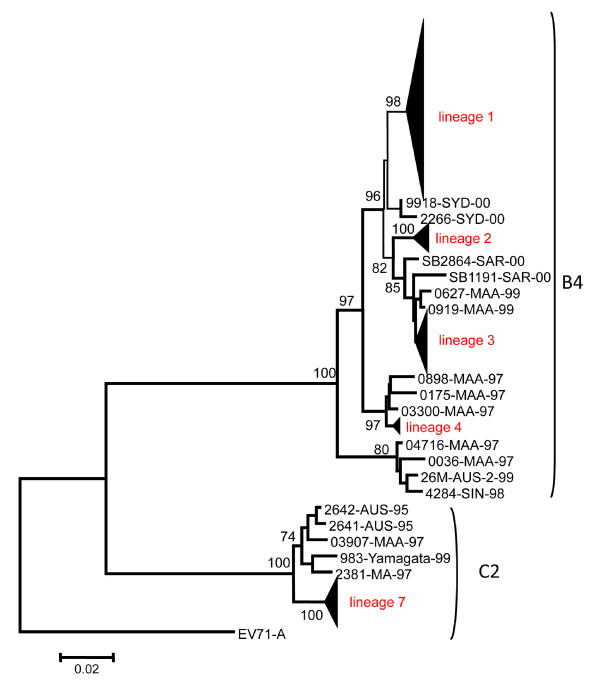
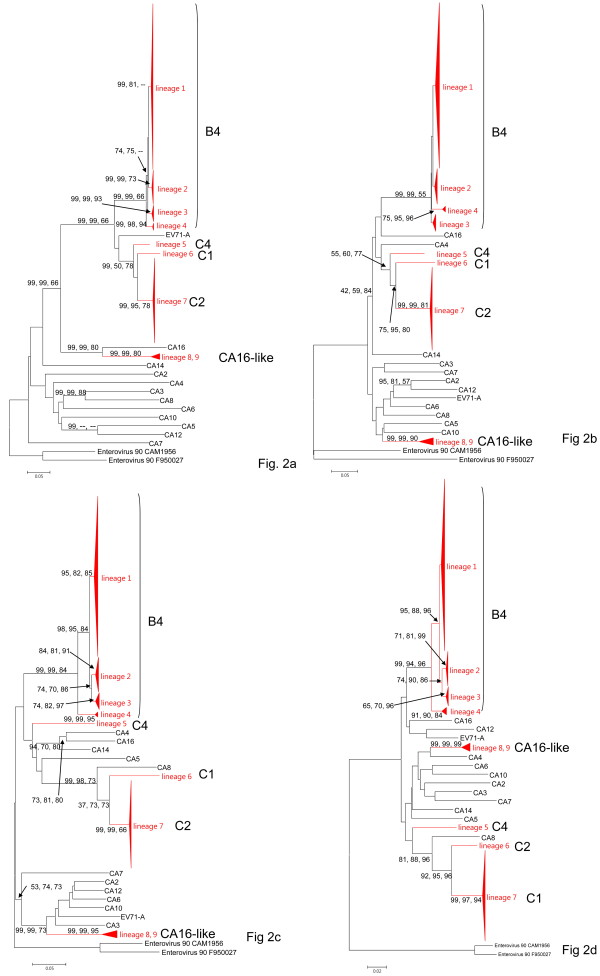
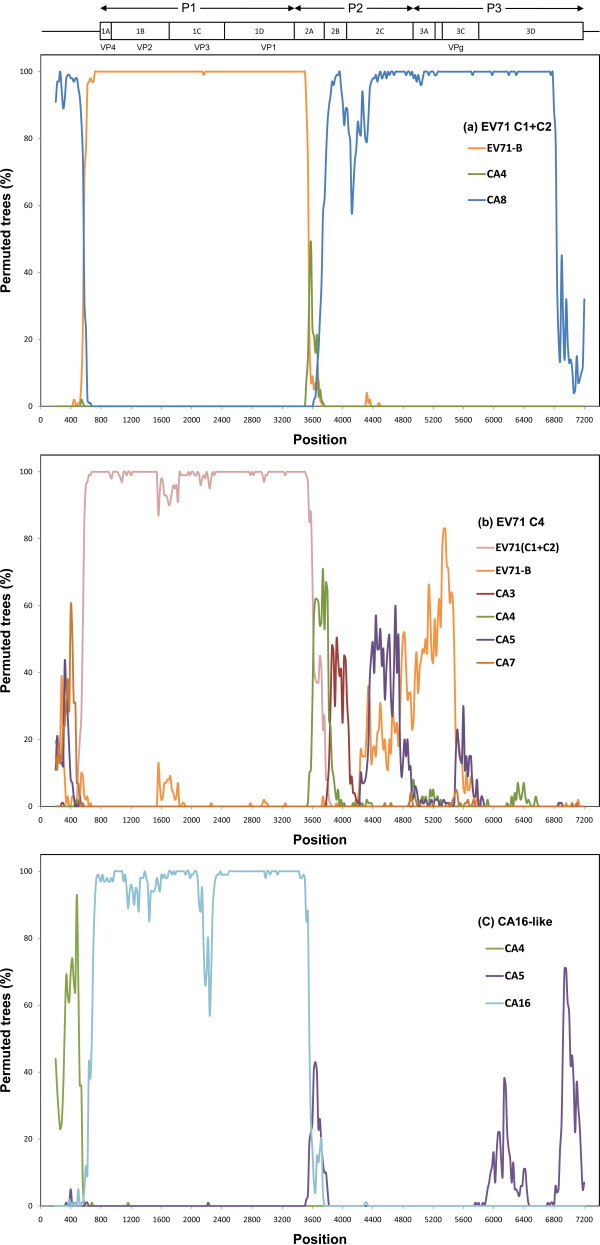
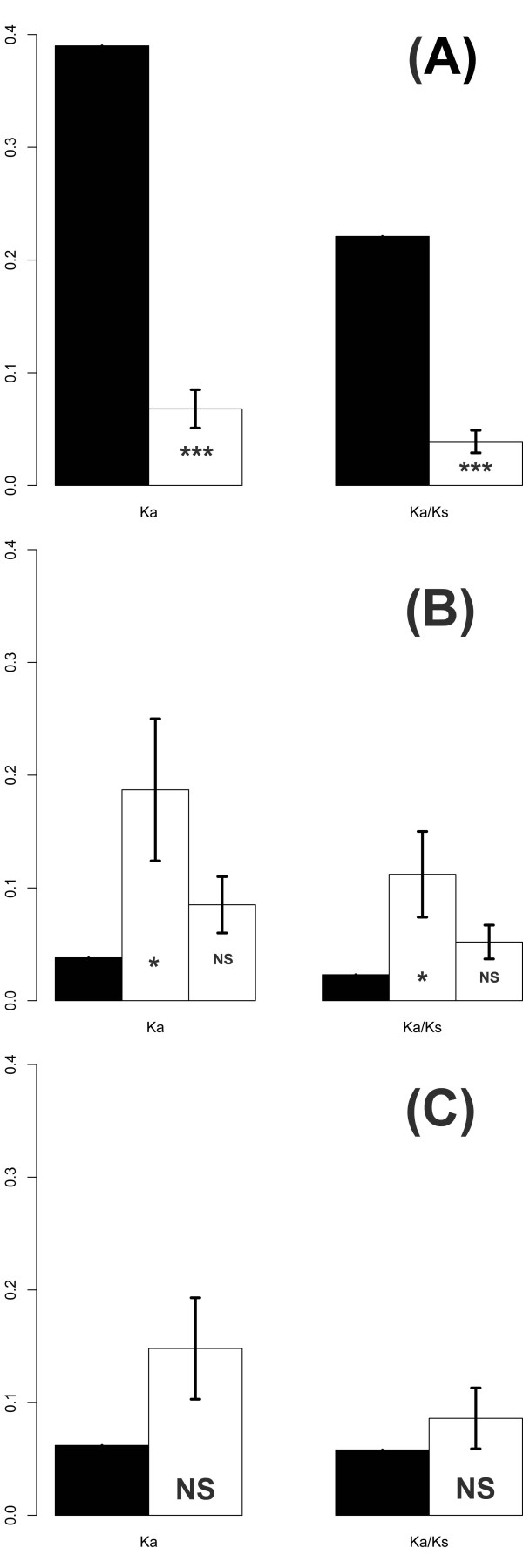
We sequenced four genomic segments, i.e., the 5' untranslated region (UTR), VP1, 2A, and 3C, of 395 EV71 viral strains collected from 1998 to 2003 in Taiwan. The phylogenies derived from different genomic segments revealed different relationships, indicating frequent sequence recombinations as previously noted. In addition to simple recombinations, exchanges of the P1 domain between different species/genotypes of human enterovirus species (HEV)-A were repeatedly observed. Contrasting patterns of polymorphisms and divergences were found between structural (VP1) and non-structural segments (2A and 3C), i.e., the former was less polymorphic within an outbreak but more divergent between different HEV-A species than the latter two. Our computer simulation demonstrated a significant excess of amino acid replacements in the VP1 region implying its possible role in adaptive evolution. Between different epidemic seasons, we observed high viral diversity in the epidemic peaks followed by severe reductions in diversity. Viruses sampled in successive epidemic seasons were not sister to each other, indicating that the annual outbreaks of EV71 were due to genetically distinct lineages.
Based on observations of accelerated amino acid changes and frequent exchanges of the P1 domain, we propose that positive selection and subsequent frequent domain shuffling are two important mechanisms for generating new genotypes of HEV-A. Our viral dynamics analysis suggested that the importation of EV71 from surrounding areas likely contributes to local EV71 outbreaks.
肠道病毒 71 型(EV71)是引起手足口病(HFMD)的常见病原体之一。近年来,该病毒引发了多次暴发疫情,导致大量死亡和严重的神经并发症。尽管这些疫情非常重要,但EV71 的进化和流行病学动态的几个方面,包括不同暴发疫情中病毒核苷酸的变异、免疫相关结构区域与非结构区域的变化率,以及推动 EV71 进化的力量,仍然不清楚。
我们对 1998 年至 2003 年间在台湾收集的 395 株 EV71 病毒株的 4 个基因组片段(5'非翻译区(UTR)、VP1、2A 和 3C)进行了测序。来自不同基因组片段的系统发育树揭示了不同的关系,表明如前所述,经常发生序列重组。除了简单重组之外,还反复观察到不同种/基因型的人类肠道病毒 A 种(HEV-A)之间 P1 结构域的交换。在结构(VP1)和非结构(2A 和 3C)区域之间发现了多态性和分歧的不同模式,即在一次暴发疫情中前者的多态性较低,但与后两者相比,不同 HEV-A 种之间的分歧较大。我们的计算机模拟表明,VP1 区域的氨基酸替换明显过剩,暗示其可能在适应性进化中起作用。在不同的流行季节之间,我们观察到在流行高峰期病毒多样性较高,随后多样性严重降低。在连续的流行季节中采样的病毒彼此之间不是姐妹关系,这表明 EV71 的年度暴发是由遗传上不同的谱系引起的。
基于观察到的加速氨基酸变化和 P1 结构域的频繁交换,我们提出正选择和随后的频繁结构域重排是产生 HEV-A 新基因型的两个重要机制。我们的病毒动力学分析表明,EV71 从周边地区的输入可能导致当地 EV71 暴发。