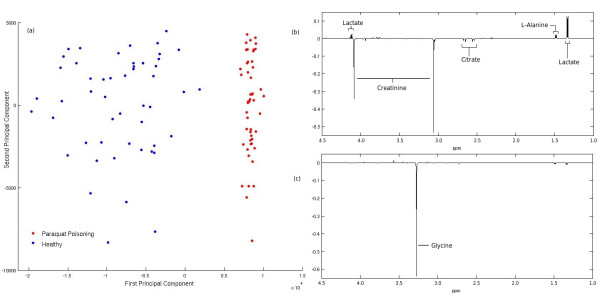
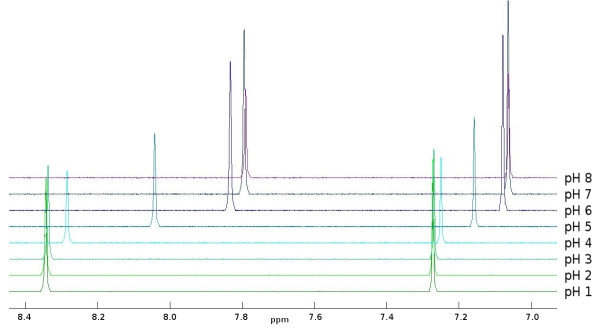
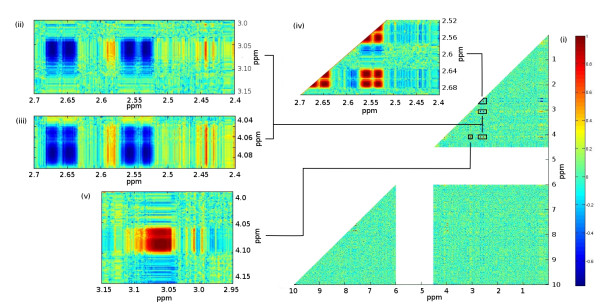
Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London, UK.
BMC Bioinformatics. 2010 Oct 6;11:496. doi: 10.1186/1471-2105-11-496.
Probing the complex fusion of genetic and environmental interactions, metabolic profiling (or metabolomics/metabonomics), the study of small molecules involved in metabolic reactions, is a rapidly expanding 'omics' field. A major technique for capturing metabolite data is 1H-NMR spectroscopy and this yields highly complex profiles that require sophisticated statistical analysis methods. However, experimental data is difficult to control and expensive to obtain. Thus data simulation is a productive route to aid algorithm development.
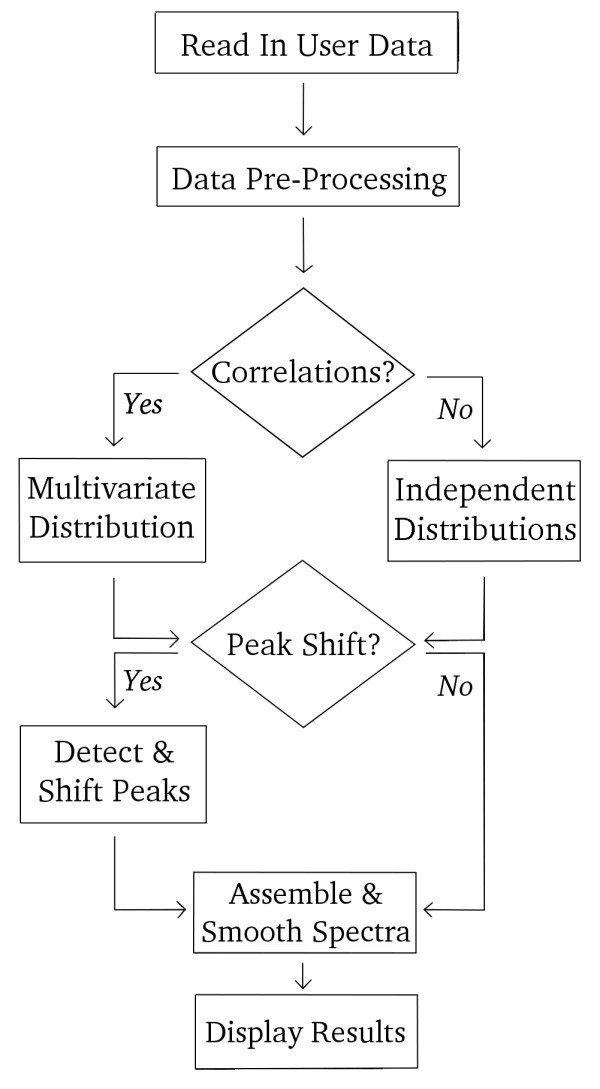
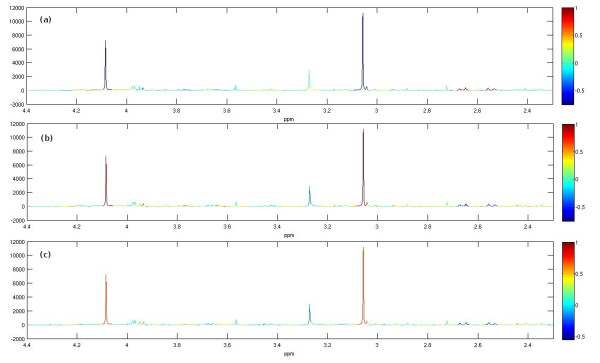
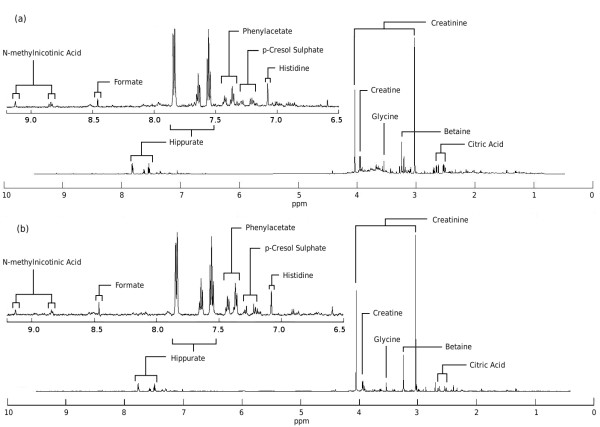
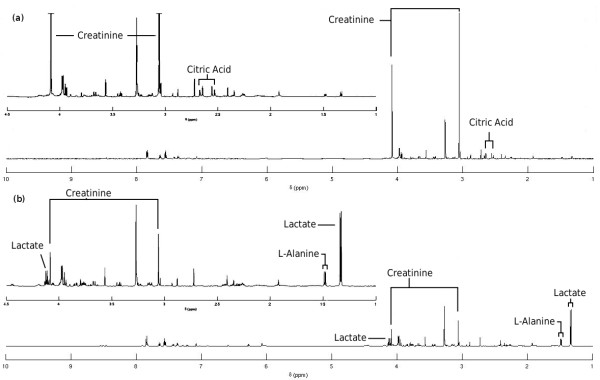
MetAssimulo is a MATLAB-based package that has been developed to simulate 1H-NMR spectra of complex mixtures such as metabolic profiles. Drawing data from a metabolite standard spectral database in conjunction with concentration information input by the user or constructed automatically from the Human Metabolome Database, MetAssimulo is able to create realistic metabolic profiles containing large numbers of metabolites with a range of user-defined properties. Current features include the simulation of two groups ('case' and 'control') specified by means and standard deviations of concentrations for each metabolite. The software enables addition of spectral noise with a realistic autocorrelation structure at user controllable levels. A crucial feature of the algorithm is its ability to simulate both intra- and inter-metabolite correlations, the analysis of which is fundamental to many techniques in the field. Further, MetAssimulo is able to simulate shifts in NMR peak positions that result from matrix effects such as pH differences which are often observed in metabolic NMR spectra and pose serious challenges for statistical algorithms.
No other software is currently able to simulate NMR metabolic profiles with such complexity and flexibility. This paper describes the algorithm behind MetAssimulo and demonstrates how it can be used to simulate realistic NMR metabolic profiles with which to develop and test new data analysis techniques. MetAssimulo is freely available for academic use at http://cisbic.bioinformatics.ic.ac.uk/metassimulo/.
代谢组学(或代谢组学/代谢组学)是研究参与代谢反应的小分子的快速发展的“组学”领域,它探索了遗传和环境相互作用的复杂融合。捕获代谢物数据的主要技术是 1H-NMR 光谱,这产生了需要复杂统计分析方法的高度复杂的图谱。然而,实验数据很难控制,获取成本也很高。因此,数据模拟是一种有前途的辅助算法开发的途径。
MetAssimulo 是一个基于 MATLAB 的软件包,用于模拟代谢物等复杂混合物的 1H-NMR 图谱。它从代谢物标准光谱数据库中提取数据,结合用户输入的浓度信息或从人类代谢物数据库中自动构建,MetAssimulo 能够创建包含大量具有用户定义属性的代谢物的现实代谢物图谱。当前的功能包括模拟两组(“病例”和“对照”),每组的浓度平均值和标准差由用户指定。该软件能够以用户可控制的水平添加具有真实自相关结构的光谱噪声。该算法的一个关键特征是能够模拟代谢物之间的内相关和相互相关,这是该领域许多技术的基础。此外,MetAssimulo 能够模拟由于 pH 差异等基质效应导致的 NMR 峰位置的移动,这些移动在代谢 NMR 光谱中经常观察到,给统计算法带来了严重的挑战。
目前没有其他软件能够以如此复杂和灵活的方式模拟 NMR 代谢物图谱。本文介绍了 MetAssimulo 背后的算法,并演示了如何使用它来模拟具有现实性的 NMR 代谢物图谱,以开发和测试新的数据分析技术。MetAssimulo 可在学术上免费使用,网址为 http://cisbic.bioinformatics.ic.ac.uk/metassimulo/。