Chen Jie, Yiğiter Ayten, Wang Yu-Ping, Deng Hong-Wen
Department of Mathematics and Statistics, University of Missouri-Kansas City, Kansas City, MO 64110, USA.
EURASIP J Bioinform Syst Biol. 2010;2010(1):268513. doi: 10.1155/2010/268513. Epub 2010 Sep 27.
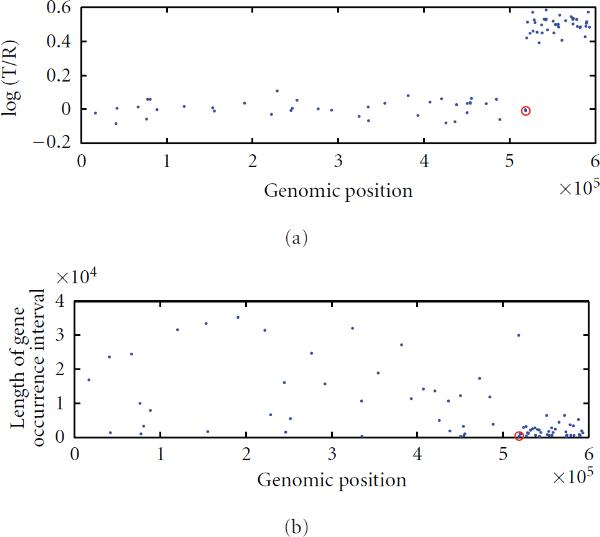
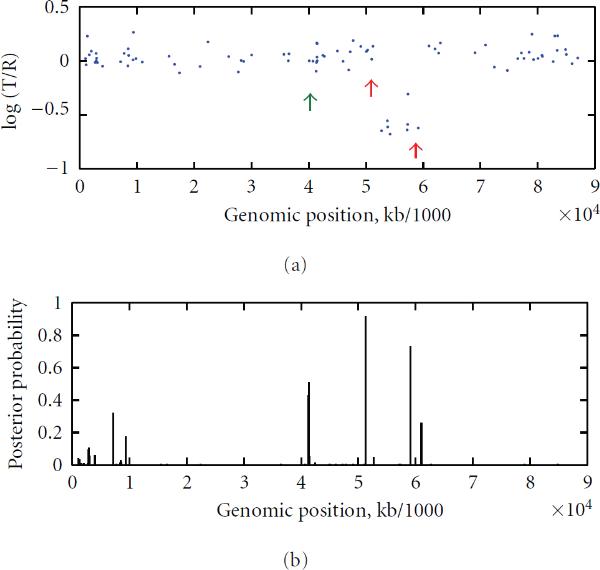
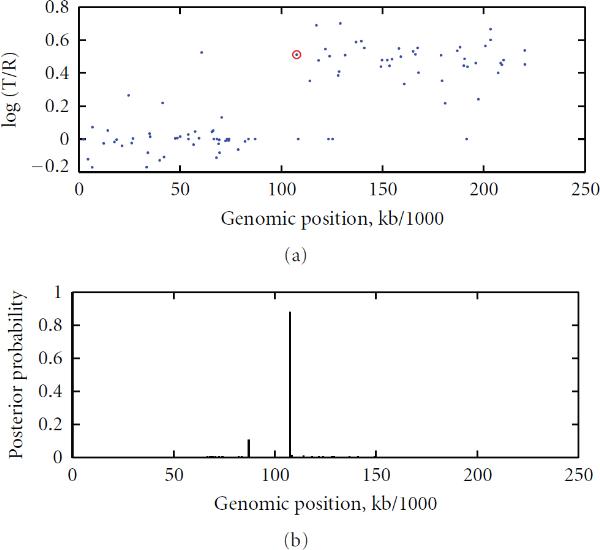
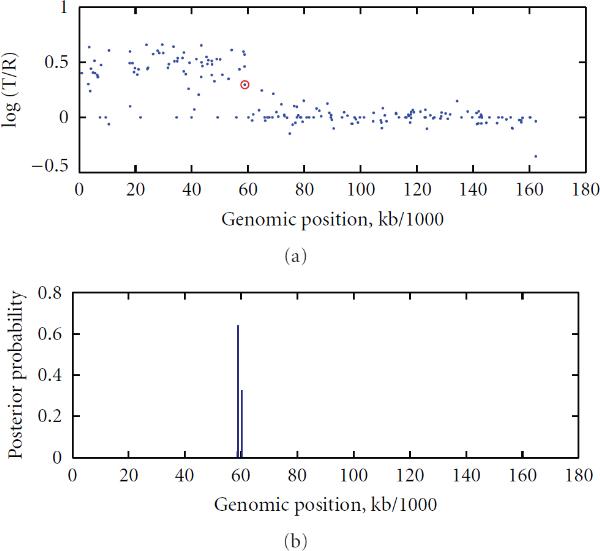
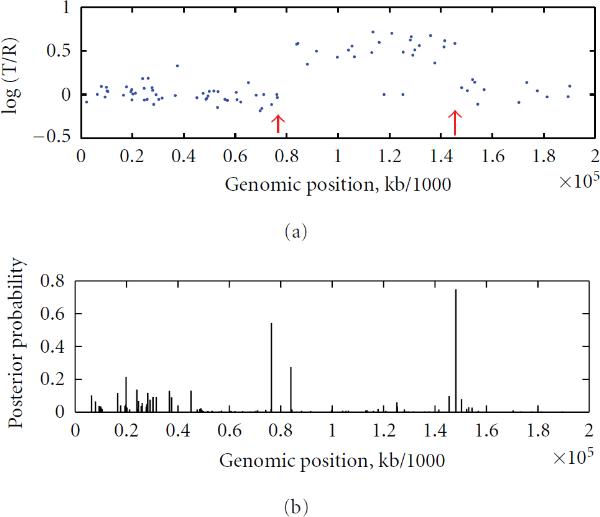
To study chromosomal aberrations that may lead to cancer formation or genetic diseases, the array-based Comparative Genomic Hybridization (aCGH) technique is often used for detecting DNA copy number variants (CNVs). Various methods have been developed for gaining CNVs information based on aCGH data. However, most of these methods make use of the log-intensity ratios in aCGH data without taking advantage of other information such as the DNA probe (e.g., biomarker) positions/distances contained in the data. Motivated by the specific features of aCGH data, we developed a novel method that takes into account the estimation of a change point or locus of the CNV in aCGH data with its associated biomarker position on the chromosome using a compound Poisson process. We used a Bayesian approach to derive the posterior probability for the estimation of the CNV locus. To detect loci of multiple CNVs in the data, a sliding window process combined with our derived Bayesian posterior probability was proposed. To evaluate the performance of the method in the estimation of the CNV locus, we first performed simulation studies. Finally, we applied our approach to real data from aCGH experiments, demonstrating its applicability.
为了研究可能导致癌症形成或遗传疾病的染色体畸变,基于阵列的比较基因组杂交(aCGH)技术常用于检测DNA拷贝数变异(CNV)。已经开发了各种方法来基于aCGH数据获取CNV信息。然而,这些方法大多利用aCGH数据中的对数强度比,而没有利用数据中包含的其他信息,如DNA探针(例如生物标志物)的位置/距离。受aCGH数据特定特征的启发,我们开发了一种新方法,该方法使用复合泊松过程,考虑aCGH数据中CNV的变化点或位点估计及其在染色体上相关生物标志物的位置。我们使用贝叶斯方法来推导估计CNV位点的后验概率。为了检测数据中多个CNV的位点,提出了一种结合我们推导的贝叶斯后验概率的滑动窗口方法。为了评估该方法在CNV位点估计中的性能,我们首先进行了模拟研究。最后,我们将我们的方法应用于aCGH实验的实际数据,证明了其适用性。