Department of Electronics and Communications Engineering, School of Information Science and Technology, Sun Yat-Sen University, 135 West Xin'gang Road, Guangzhou, P.R. China.
Evol Bioinform Online. 2010 Sep 20;6:113-31. doi: 10.4137/ebo.s5602.
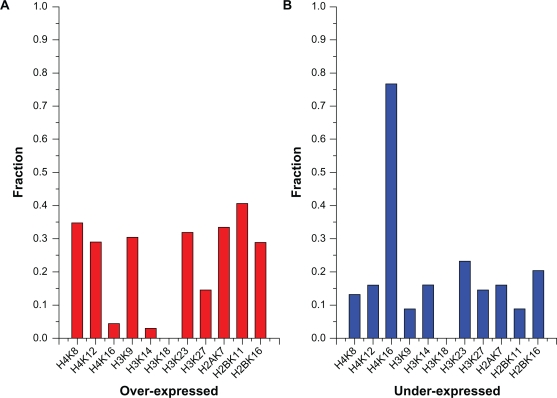
Eukaryotic genomes are packaged into chromatin by histone proteins whose chemical modification can profoundly influence gene expression. The histone modifications often act in combinations, which exert different effects on gene expression. Although a number of experimental techniques and data analysis methods have been developed to study histone modifications, it is still very difficult to identify the relationships among histone modifications on a genome-wide scale.We proposed a method to identify the combinatorial effects of histone modifications by association rule mining. The method first identified Functional Modification Transactions (FMTs) and then employed association rule mining algorithm and statistics methods to identify histone modification patterns. We applied the proposed methodology to Pokholok et al's data with eight sets of histone modifications and Kurdistani et al's data with eleven histone acetylation sites. Our method succeeds in revealing two different global views of histone modification landscapes on two datasets and identifying a number of modification patterns some of which are supported by previous studies.We concentrate on combinatorial effects of histone modifications which significantly affect gene expression. Our method succeeds in identifying known interactions among histone modifications and uncovering many previously unknown patterns. After in-depth analysis of possible mechanism by which histone modification patterns can alter transcriptional states, we infer three possible modification pattern reading mechanism ('redundant', 'trivial', 'dominative'). Our results demonstrate several histone modification patterns which show significant correspondence between yeast and human cells.
真核生物基因组通过组蛋白蛋白包装成染色质,组蛋白蛋白的化学修饰可以深刻影响基因表达。组蛋白修饰通常以组合的形式发挥作用,对基因表达产生不同的影响。尽管已经开发了许多实验技术和数据分析方法来研究组蛋白修饰,但要在全基因组范围内识别组蛋白修饰之间的关系仍然非常困难。
我们提出了一种通过关联规则挖掘来识别组蛋白修饰组合效应的方法。该方法首先识别功能修饰转换(FMT),然后采用关联规则挖掘算法和统计方法来识别组蛋白修饰模式。我们将所提出的方法应用于 Pokholok 等人的包含 8 组组蛋白修饰的数据和 Kurdistani 等人的包含 11 个组蛋白乙酰化位点的数据。我们的方法成功地揭示了两个数据集上组蛋白修饰景观的两种不同全局视图,并识别了许多修饰模式,其中一些得到了先前研究的支持。
我们专注于显著影响基因表达的组蛋白修饰的组合效应。我们的方法成功地识别了组蛋白修饰之间已知的相互作用,并揭示了许多以前未知的模式。在深入分析组蛋白修饰模式如何改变转录状态的可能机制后,我们推断了三种可能的修饰模式读取机制(“冗余”、“琐碎”、“主导”)。我们的结果表明,酵母和人类细胞之间存在几种组蛋白修饰模式,它们之间存在显著的一致性。