Center for Statistical Sciences and Department of Community Health, Box G-121S-7, Brown University, Providence RI 02912, USA.
BMC Bioinformatics. 2010 Nov 16;11:564. doi: 10.1186/1471-2105-11-564.
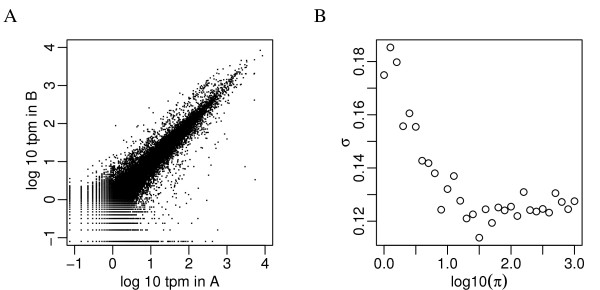
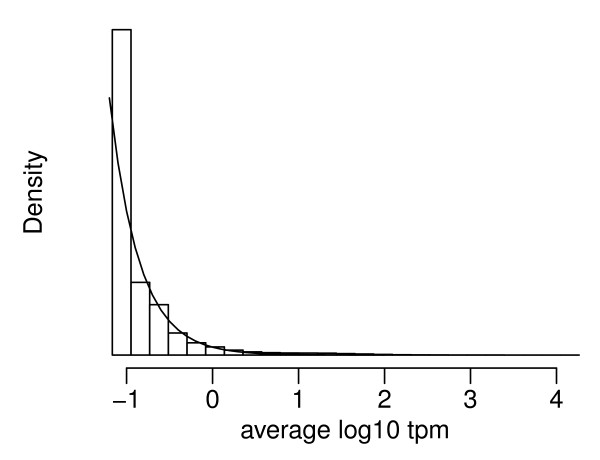
Recent technological advancements have made high throughput sequencing an increasingly popular approach for transcriptome analysis. Advantages of sequencing-based transcriptional profiling over microarrays have been reported, including lower technical variability. However, advances in technology do not remove biological variation between replicates and this variation is often neglected in many analyses.
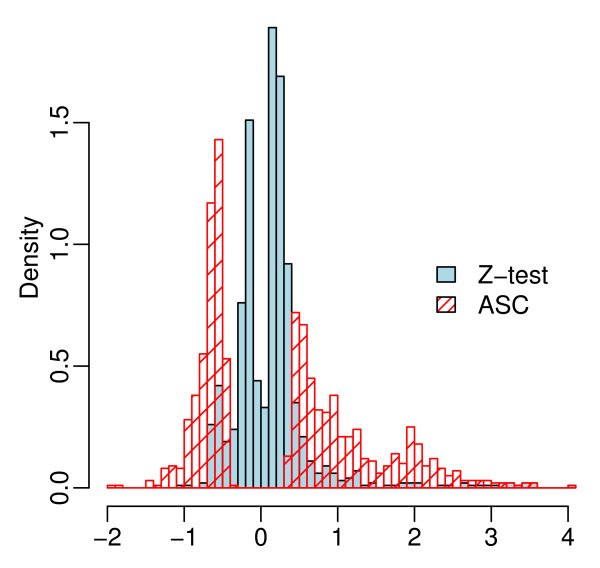
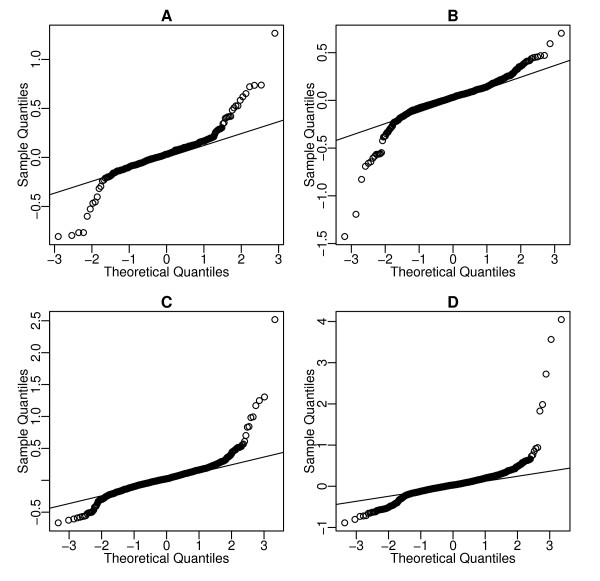
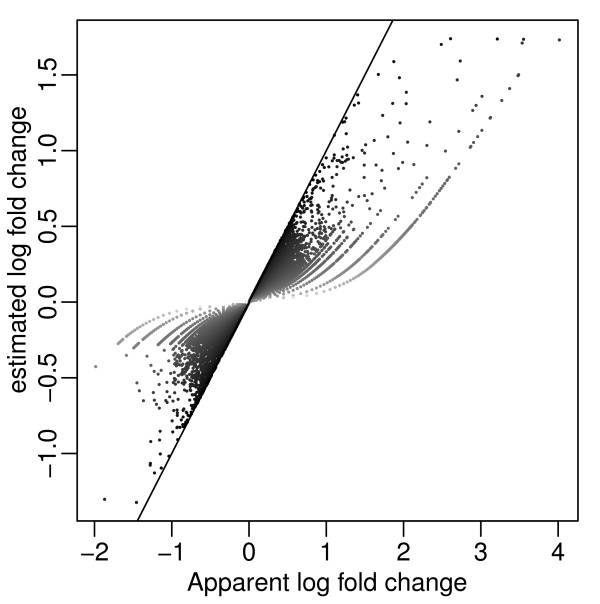
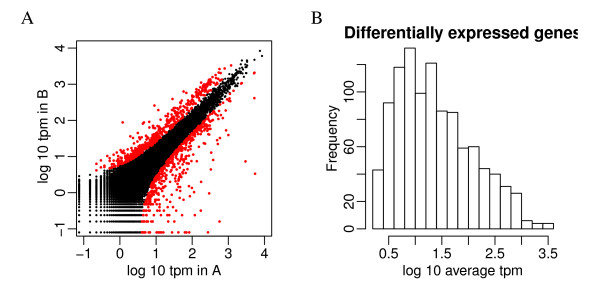
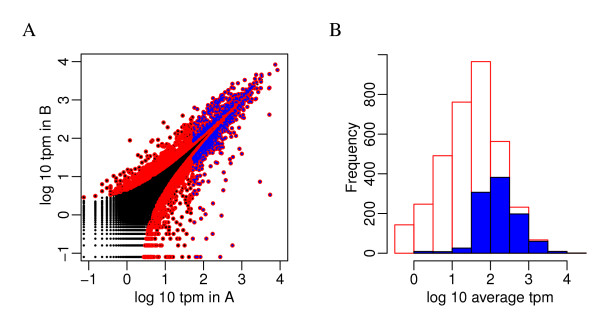
We propose an empirical Bayes method, titled Analysis of Sequence Counts (ASC), to detect differential expression based on sequencing technology. ASC borrows information across sequences to establish prior distribution of sample variation, so that biological variation can be accounted for even when replicates are not available. Compared to current approaches that simply tests for equality of proportions in two samples, ASC is less biased towards highly expressed sequences and can identify more genes with a greater log fold change at lower overall abundance.
ASC unifies the biological and statistical significance of differential expression by estimating the posterior mean of log fold change and estimating false discovery rates based on the posterior mean. The implementation in R is available at http://www.stat.brown.edu/Zwu/research.aspx.
最近的技术进步使得高通量测序成为转录组分析越来越受欢迎的方法。与微阵列相比,基于测序的转录组分析具有技术变异性较低等优势。然而,技术的进步并没有消除重复之间的生物学变异性,而许多分析往往忽略了这种变异性。
我们提出了一种经验贝叶斯方法,称为序列计数分析(ASC),用于基于测序技术检测差异表达。ASC 通过跨序列借用信息来建立样本变异的先验分布,因此即使没有重复样本,也可以考虑生物学变异。与当前仅测试两个样本中比例是否相等的方法相比,ASC 对高表达序列的偏差较小,并且可以在整体丰度较低的情况下识别更多具有更大对数倍数变化的基因。
ASC 通过估计对数倍数变化的后验均值,并基于后验均值估计错误发现率,从而统一了差异表达的生物学和统计学意义。R 中的实现可在 http://www.stat.brown.edu/Zwu/research.aspx 获得。