Department of Electrical Engineering, University of Texas at San Antonio, Texas, USA.
BMC Genomics. 2010 Dec 1;11 Suppl 3(Suppl 3):S8. doi: 10.1186/1471-2164-11-S3-S8.
The identification and quantification of proteins using label-free Liquid Chromatography/Mass Spectrometry (LC/MS) play crucial roles in biological and biomedical research. Increasing evidence has shown that biomarkers are often low abundance proteins. However, LC/MS systems are subject to considerable noise and sample variability, whose statistical characteristics are still elusive, making computational identification of low abundance proteins extremely challenging. As a result, the inability of identifying low abundance proteins in a proteomic study is the main bottleneck in protein biomarker discovery.
In this paper, we propose a new peak detection method called Information Combining Peak Detection (ICPD ) for high resolution LC/MS. In LC/MS, peptides elute during a certain time period and as a result, peptide isotope patterns are registered in multiple MS scans. The key feature of the new algorithm is that the observed isotope patterns registered in multiple scans are combined together for estimating the likelihood of the peptide existence. An isotope pattern matching score based on the likelihood probability is provided and utilized for peak detection.
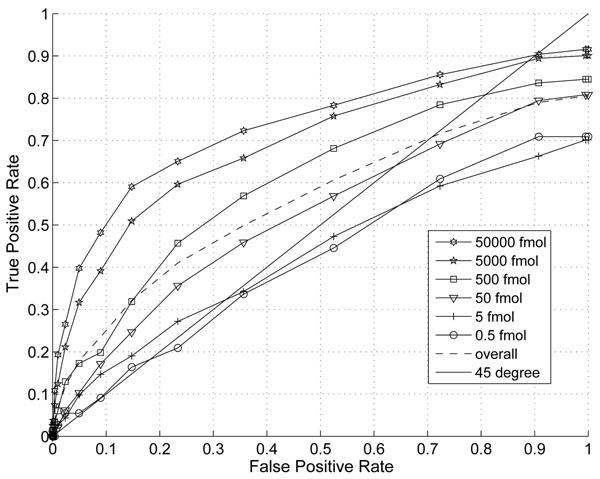
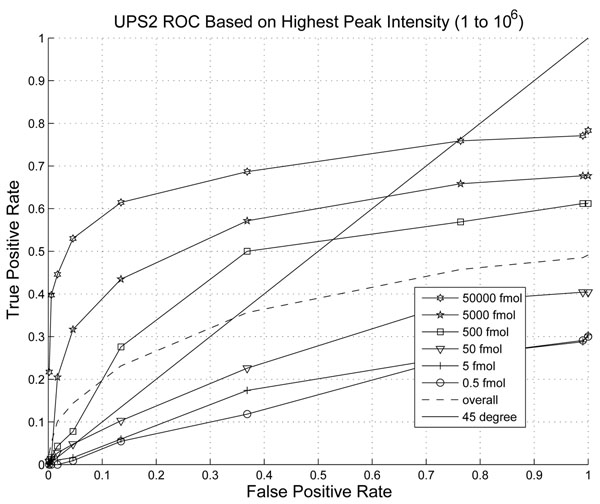
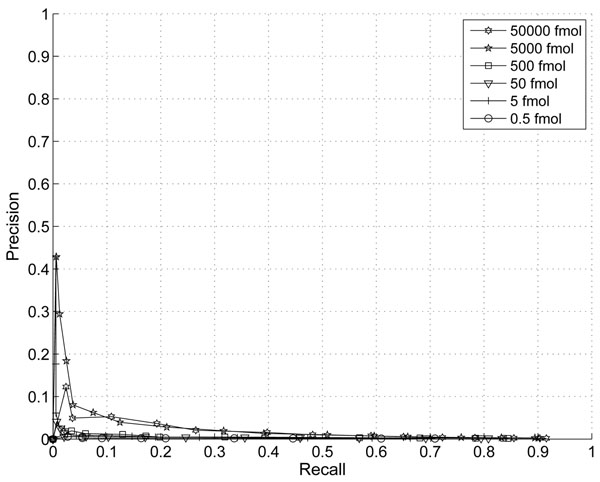
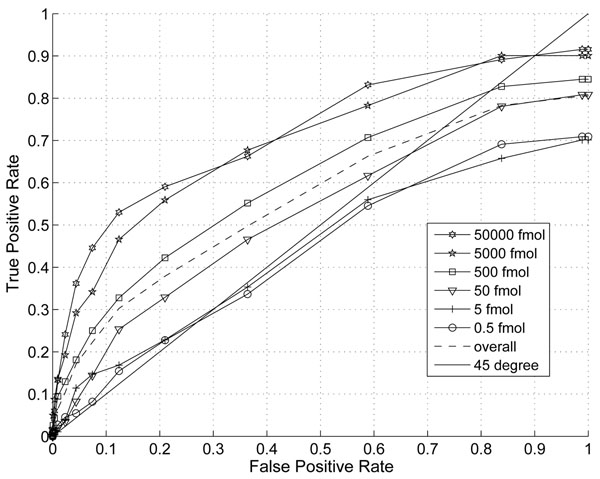
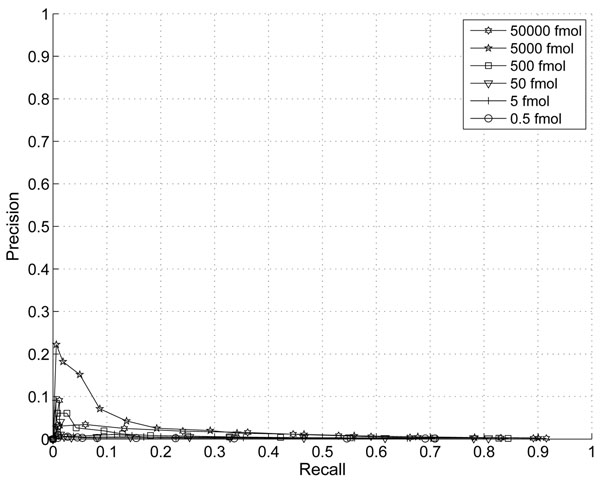
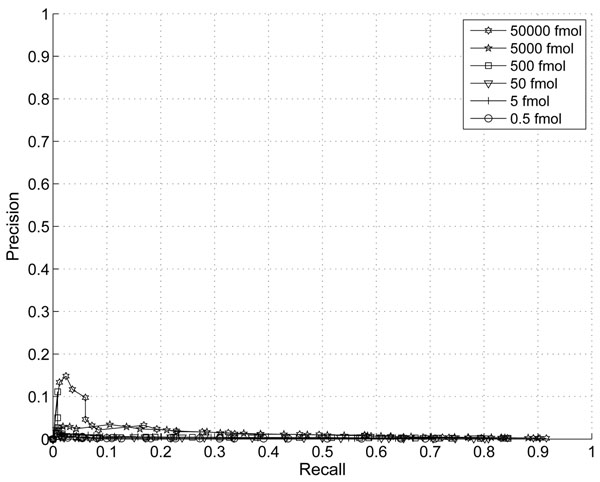
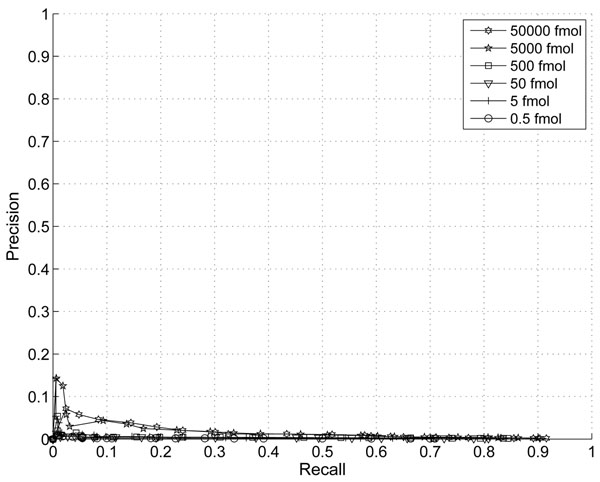
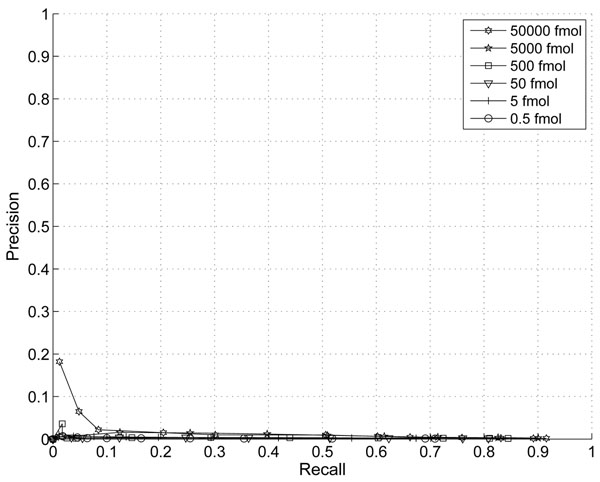
The performance of the new algorithm is evaluated based on protein standards with 48 known proteins. The evaluation shows better peak detection accuracy for low abundance proteins than other LC/MS peak detection methods.
使用无标记液相色谱/质谱(LC/MS)鉴定和定量蛋白质在生物和生物医学研究中起着至关重要的作用。越来越多的证据表明,生物标志物通常是低丰度蛋白质。然而,LC/MS 系统受到相当大的噪声和样品可变性的影响,其统计特征仍然难以捉摸,这使得低丰度蛋白质的计算鉴定极具挑战性。因此,在蛋白质组学研究中无法识别低丰度蛋白质是蛋白质生物标志物发现的主要瓶颈。
在本文中,我们提出了一种新的峰检测方法,称为信息组合峰检测(ICPD),用于高分辨率 LC/MS。在 LC/MS 中,肽在特定时间段内洗脱,因此肽同位素模式在多个 MS 扫描中注册。新算法的关键特征是,将在多个扫描中注册的观察到的同位素模式组合在一起,以估计肽存在的可能性。基于似然概率提供并利用同位素模式匹配得分进行峰检测。
基于具有 48 种已知蛋白质的蛋白质标准品评估了新算法的性能。评估结果表明,与其他 LC/MS 峰检测方法相比,该新算法对低丰度蛋白质具有更好的峰检测准确性。