Department of Electrical and Computer Engineering, the University of Texas at San Antonio, One UTSA Circle, San Antonio, TX 78249, USA.
BMC Bioinformatics. 2011 Nov 11;12:439. doi: 10.1186/1471-2105-12-439.
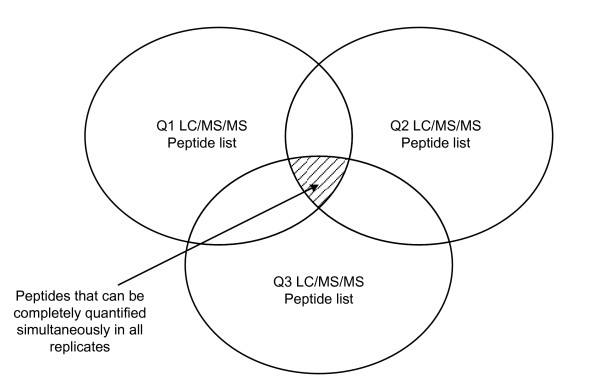
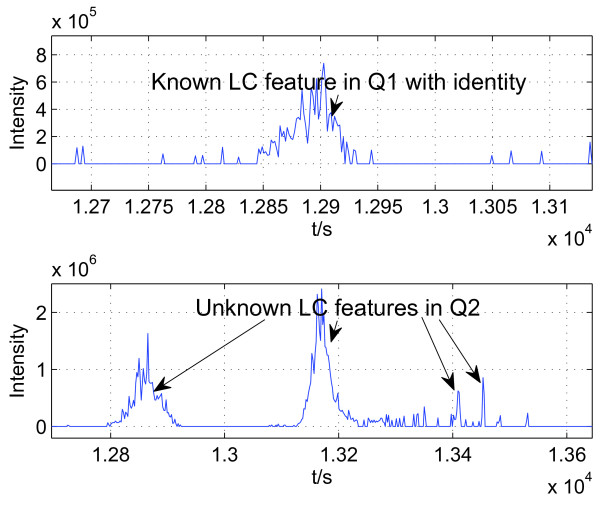
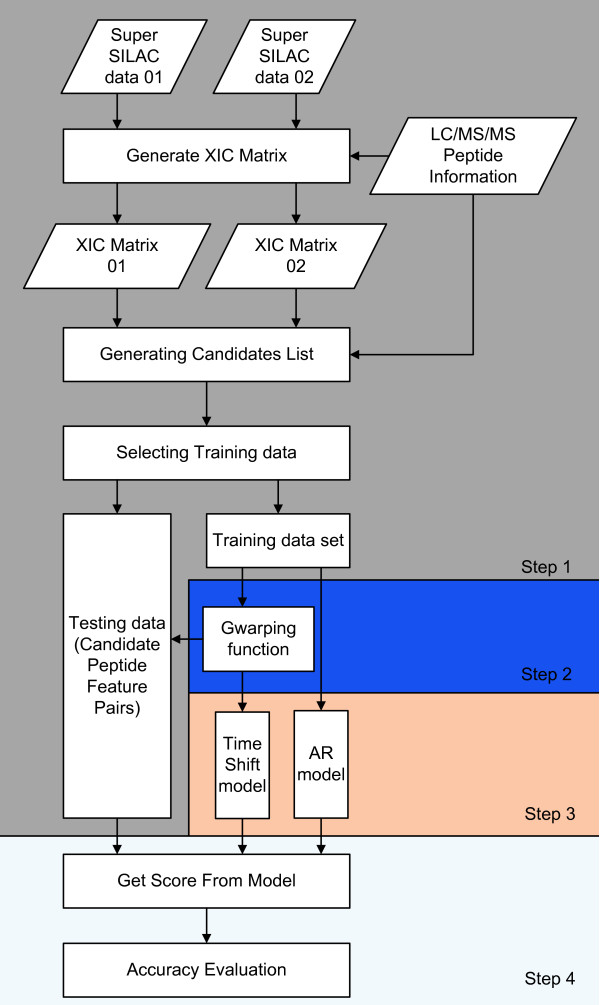
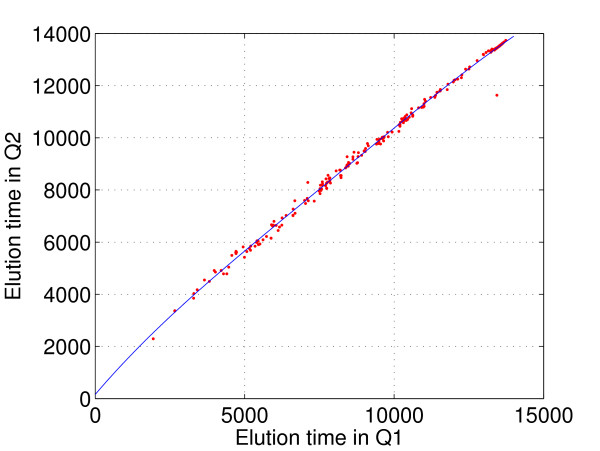
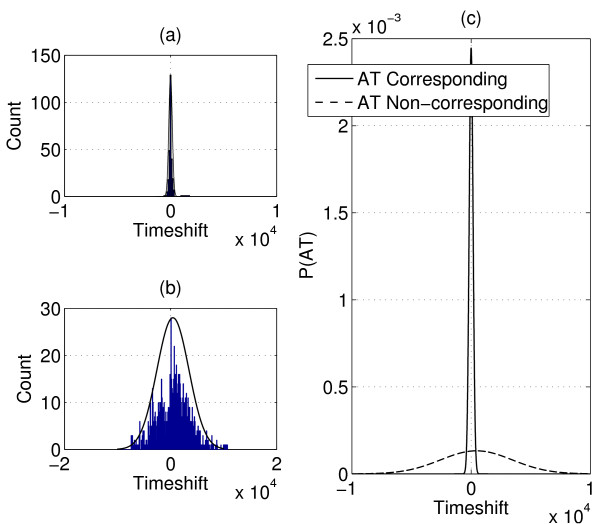
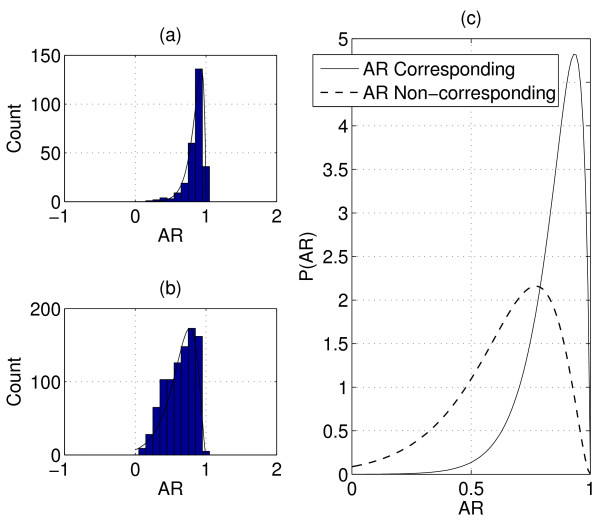
Identifying corresponding features (LC peaks registered by identical peptides) in multiple Liquid Chromatography/Mass Spectrometry (LC-MS) datasets plays a crucial role in the analysis of complex peptide or protein mixtures. Warping functions are commonly used to correct the mean of elution time shifts among LC-MS datasets, which cannot resolve the ambiguity of corresponding feature identification since elution time shifts are random. We propose a Statistical Corresponding Feature Identification Algorithm(SCFIA) based on both elution time shifts and peak shape correlations between corresponding features. SCFIA first trains a set of statistical models, and then, all candidate corresponding features are scored by the statistical models to find the maximum likelihood solution.
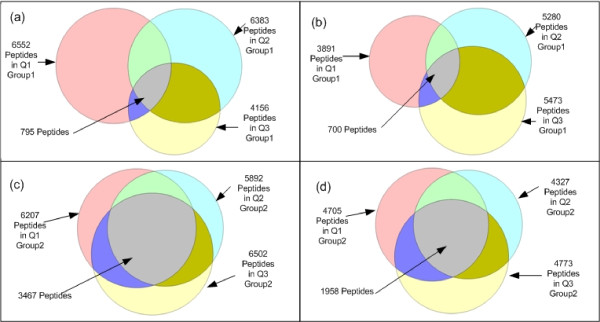
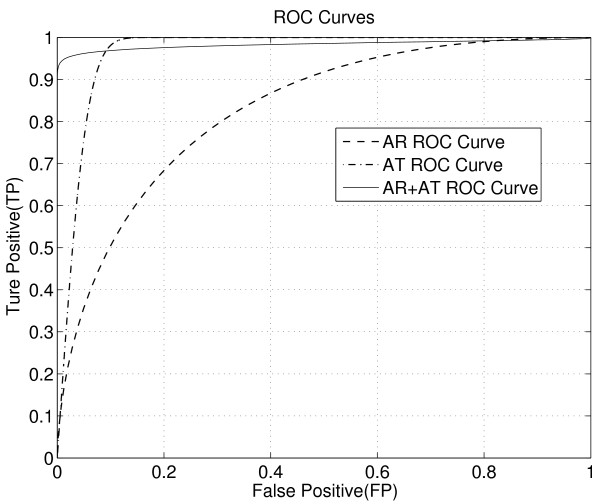
We test SCFIA on publicly available datasets. We first compare its performance with that of warping function based methods, and the results show significant improvements. The performance of SCFIA on replicates datasets and fractionated datasets is also evaluated. In both cases, the accuracy is above 90%, which is near optimal. Finally the coverage of SCFIA is evaluated, and it is shown that SCFIA can find corresponding features in multiple datasets for over 90% peptides identified by Tandem MS.
SCFIA can be used for accurate corresponding feature identification in LC-MS. We have shown that peak shape correlation can be used effectively for improving the accuracy. SCFIA provides high coverage in corresponding feature identification in multiple datasets, which serves the basis for integrating multiple LC-MS measurements for accurate peptide quantification.
在分析复杂的肽或蛋白质混合物时,识别多个液相色谱/质谱(LC-MS)数据集之间的对应特征(通过相同的肽注册的 LC 峰)至关重要。扭曲函数通常用于校正 LC-MS 数据集之间的洗脱时间偏移的平均值,但由于洗脱时间偏移是随机的,因此无法解决对应特征识别的模糊性。我们提出了一种基于洗脱时间偏移和对应特征之间的峰形相关性的统计对应特征识别算法(SCFIA)。SCFIA 首先训练一组统计模型,然后通过统计模型对所有候选对应特征进行评分,以找到最大似然解。
我们在公开可用的数据集上测试了 SCFIA。我们首先将其性能与基于扭曲函数的方法进行比较,结果表明有显著的改进。还评估了 SCFIA 在重复数据集和分馏数据集上的性能。在这两种情况下,准确率都在 90%以上,接近最优。最后评估了 SCFIA 的覆盖范围,结果表明 SCFIA 可以在多个数据集为超过 90%的通过串联 MS 鉴定的肽找到对应特征。
SCFIA 可用于 LC-MS 中准确的对应特征识别。我们已经表明,峰形相关性可有效用于提高准确性。SCFIA 在多个数据集的对应特征识别中具有较高的覆盖率,为准确的肽定量整合多个 LC-MS 测量提供了基础。