Department of Biological and Biomedical Sciences, Interdepartmental Program in Computational Biology and Bioinformatics, Yale University School of Medicine, 310 Cedar Street, New Haven, CT 06520, USA.
Bioinformatics. 2011 Feb 1;27(3):416-8. doi: 10.1093/bioinformatics/btq658. Epub 2010 Dec 12.
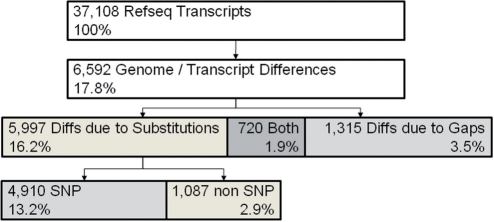
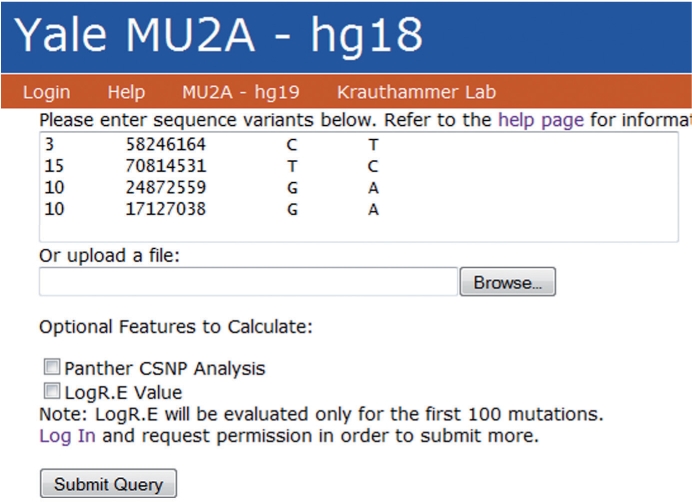
Next-generation sequencing technologies enable the identification of sequence variation in the genome and transcriptome. Differences between the reference genome and transcript libraries complicate the determination of the effect of genomic sequence variants on protein products; similarly, these differences complicate the mapping of sequence variants found in transcripts to their respective genomic position. We have developed MU2A, a publicly available web service for variant annotation that reconciles differences between the genome and transcriptome, enabling the rapid and accurate determination of the effects of genomic variants on protein products, and the mapping of variants detected in transcripts to genomic coordinates. The MU2A web service is available at http://krauthammerlab.med.yale.edu/mu2a. We have released MU2A as open source, available at http://code.google.com/p/mu2a/.
下一代测序技术使我们能够鉴定基因组和转录组中的序列变异。参考基因组和转录文库之间的差异使得确定基因组序列变异对蛋白质产物的影响变得复杂;同样,这些差异也使得将在转录本中发现的序列变异映射到其各自的基因组位置变得复杂。我们开发了 MU2A,这是一个公开可用的变体注释的网络服务,它可以协调基因组和转录组之间的差异,从而快速准确地确定基因组变异对蛋白质产物的影响,并将在转录本中检测到的变异映射到基因组坐标上。MU2A 网络服务可在 http://krauthammerlab.med.yale.edu/mu2a 上获得。我们已经将 MU2A 作为开源软件发布,可在 http://code.google.com/p/mu2a/ 上获得。