Clayton School of Information Technology, Monash University, Clayton 3800, Australia.
BMC Bioinformatics. 2010 Dec 16;11:599. doi: 10.1186/1471-2105-11-599.
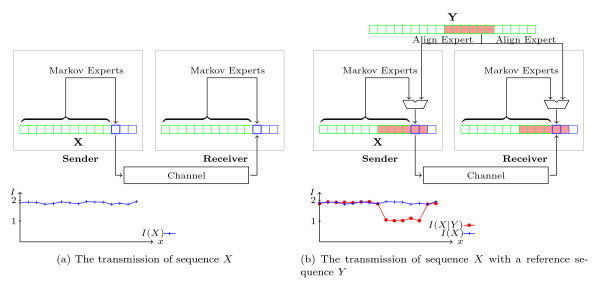
Traditional genome alignment methods consider sequence alignment as a variation of the string edit distance problem, and perform alignment by matching characters of the two sequences. They are often computationally expensive and unable to deal with low information regions. Furthermore, they lack a well-principled objective function to measure the performance of sets of parameters. Since genomic sequences carry genetic information, this article proposes that the information content of each nucleotide in a position should be considered in sequence alignment. An information-theoretic approach for pairwise genome local alignment, namely XMAligner, is presented. Instead of comparing sequences at the character level, XMAligner considers a pair of nucleotides from two sequences to be related if their mutual information in context is significant. The information content of nucleotides in sequences is measured by a lossless compression technique.
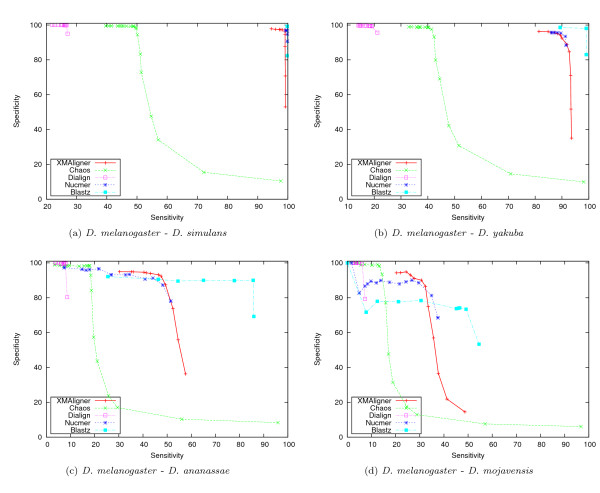
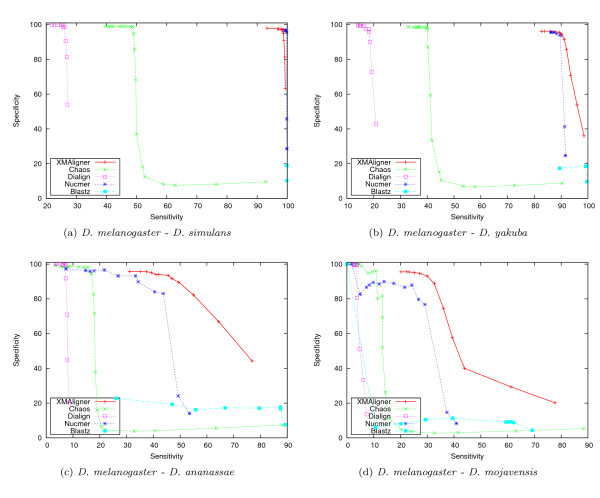
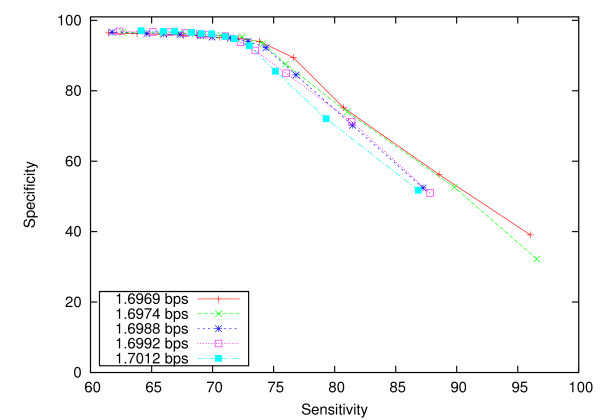
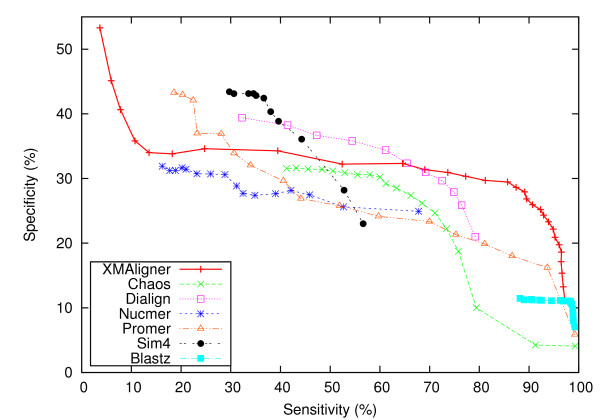
Experiments on both simulated data and real data show that XMAligner is superior to conventional methods especially on distantly related sequences and statistically biased data. XMAligner can align sequences of eukaryote genome size with only a modest hardware requirement. Importantly, the method has an objective function which can obviate the need to choose parameter values for high quality alignment. The alignment results from XMAligner can be integrated into a visualisation tool for viewing purpose.
The information-theoretic approach for sequence alignment is shown to overcome the mentioned problems of conventional character matching alignment methods. The article shows that, as genomic sequences are meant to carry information, considering the information content of nucleotides is helpful for genomic sequence alignment.
Downloadable binaries, documentation and data can be found at ftp://ftp.infotech.monash.edu.au/software/DNAcompress-XM/XMAligner/.
传统的基因组比对方法将序列比对视为字符串编辑距离问题的变体,并通过匹配两个序列的字符来进行比对。它们通常计算成本高昂,并且无法处理低信息区域。此外,它们缺乏一种良好的有原则的目标函数来衡量参数集的性能。由于基因组序列携带遗传信息,本文提出在序列比对中应考虑每个位置核苷酸的信息含量。提出了一种用于成对基因组局部比对的信息论方法,即 XMAligner。XMAligner 不是在字符级别比较序列,而是考虑如果两个序列中一对核苷酸的上下文互信息显著,则它们相关。序列中核苷酸的信息含量通过无损压缩技术来衡量。
在模拟数据和真实数据上的实验表明,XMAligner 优于传统方法,尤其是在远缘序列和统计偏差数据上。XMAligner 可以仅使用适度的硬件要求对齐真核生物基因组大小的序列。重要的是,该方法具有一个目标函数,可以避免为高质量对齐选择参数值的需要。XMAligner 的对齐结果可以集成到可视化工具中用于查看。
序列比对的信息论方法被证明可以克服传统字符匹配比对方法的所述问题。本文表明,由于基因组序列旨在携带信息,因此考虑核苷酸的信息含量有助于基因组序列比对。
可下载的二进制文件、文档和数据可在 ftp://ftp.infotech.monash.edu.au/software/DNAcompress-XM/XMAligner/ 找到。