Signal Processing Lab, IEETA/DETI, University of Aveiro, Aveiro, Portugal.
PLoS One. 2011;6(6):e21588. doi: 10.1371/journal.pone.0021588. Epub 2011 Jun 30.
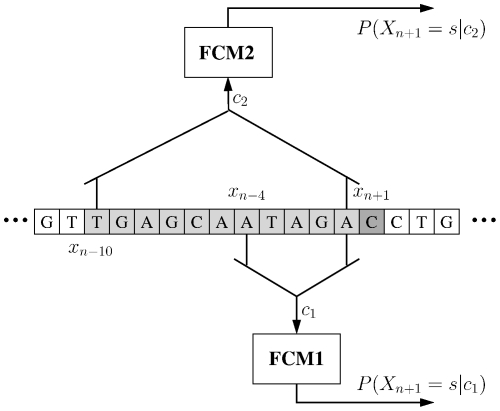
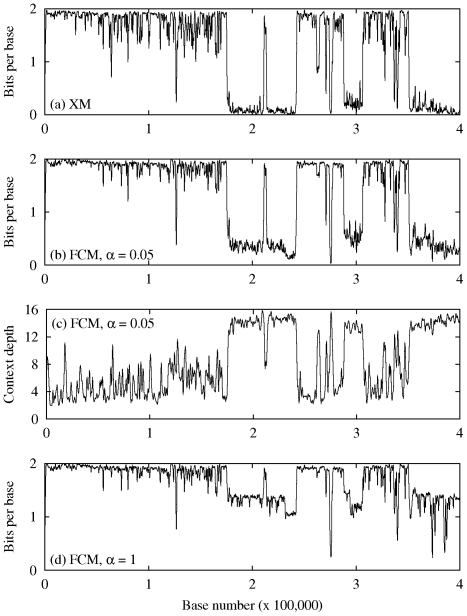
A finite-context (Markov) model of order k yields the probability distribution of the next symbol in a sequence of symbols, given the recent past up to depth k. Markov modeling has long been applied to DNA sequences, for example to find gene-coding regions. With the first studies came the discovery that DNA sequences are non-stationary: distinct regions require distinct model orders. Since then, Markov and hidden Markov models have been extensively used to describe the gene structure of prokaryotes and eukaryotes. However, to our knowledge, a comprehensive study about the potential of Markov models to describe complete genomes is still lacking. We address this gap in this paper. Our approach relies on (i) multiple competing Markov models of different orders (ii) careful programming techniques that allow orders as large as sixteen (iii) adequate inverted repeat handling (iv) probability estimates suited to the wide range of context depths used. To measure how well a model fits the data at a particular position in the sequence we use the negative logarithm of the probability estimate at that position. The measure yields information profiles of the sequence, which are of independent interest. The average over the entire sequence, which amounts to the average number of bits per base needed to describe the sequence, is used as a global performance measure. Our main conclusion is that, from the probabilistic or information theoretic point of view and according to this performance measure, multiple competing Markov models explain entire genomes almost as well or even better than state-of-the-art DNA compression methods, such as XM, which rely on very different statistical models. This is surprising, because Markov models are local (short-range), contrasting with the statistical models underlying other methods, where the extensive data repetitions in DNA sequences is explored, and therefore have a non-local character.
一种有限上下文(马尔可夫)模型的阶 k 给出了符号序列中下一个符号的概率分布,给定最近过去的深度 k。马尔可夫模型长期以来一直应用于 DNA 序列,例如用于找到基因编码区域。随着第一批研究的出现,人们发现 DNA 序列是非平稳的:不同的区域需要不同的模型阶。从那时起,马尔可夫和隐马尔可夫模型被广泛用于描述原核生物和真核生物的基因结构。然而,据我们所知,关于马尔可夫模型描述完整基因组的潜力的综合研究仍然缺乏。我们在本文中解决了这一差距。我们的方法依赖于(i)不同阶的多个竞争马尔可夫模型(ii)允许阶数高达十六的精心编程技术(iii)适当的反转重复处理(iv)适合使用的广泛上下文深度的概率估计。为了衡量模型在序列中特定位置拟合数据的程度,我们使用该位置概率估计的负对数。该度量给出了序列的信息概况,这些概况具有独立的意义。整个序列的平均值,即描述序列所需的平均每个碱基的位数,用作全局性能度量。我们的主要结论是,从概率或信息理论的角度来看,并且根据这个性能度量,多个竞争的马尔可夫模型几乎可以与最先进的 DNA 压缩方法(例如 XM)一样或更好地解释整个基因组,后者依赖于非常不同的统计模型。这令人惊讶,因为马尔可夫模型是局部的(短程的),与其他方法的统计模型形成对比,其他方法探索了 DNA 序列中的广泛数据重复,因此具有非局部特征。