Institute of Electronics and Informatics Engineering of Aveiro, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal.
Department of Electronics Telecommunications and Informatics, University of Aveiro, Campus Universitario de Santiago, 3810-193 Aveiro, Portugal.
Gigascience. 2022 Aug 11;11. doi: 10.1093/gigascience/giac079.
Viruses are among the shortest yet highly abundant species that harbor minimal instructions to infect cells, adapt, multiply, and exist. However, with the current substantial availability of viral genome sequences, the scientific repertory lacks a complexity landscape that automatically enlights viral genomes' organization, relation, and fundamental characteristics.
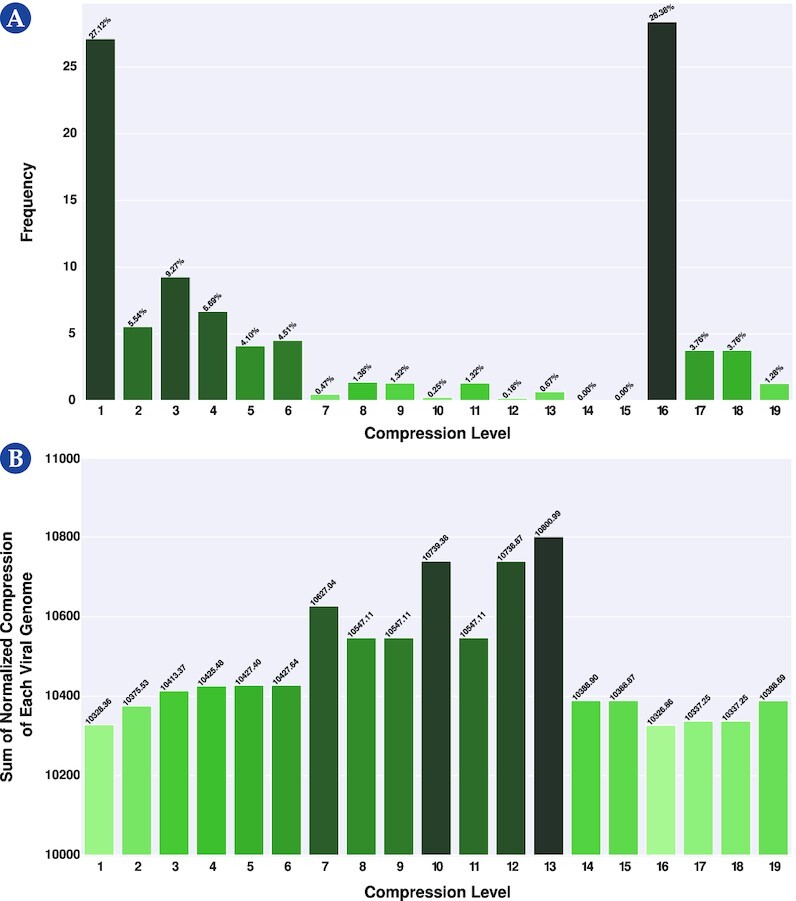
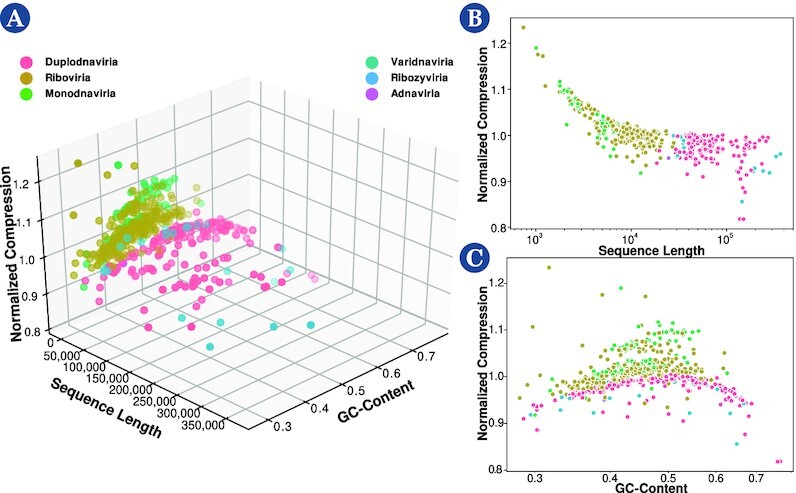
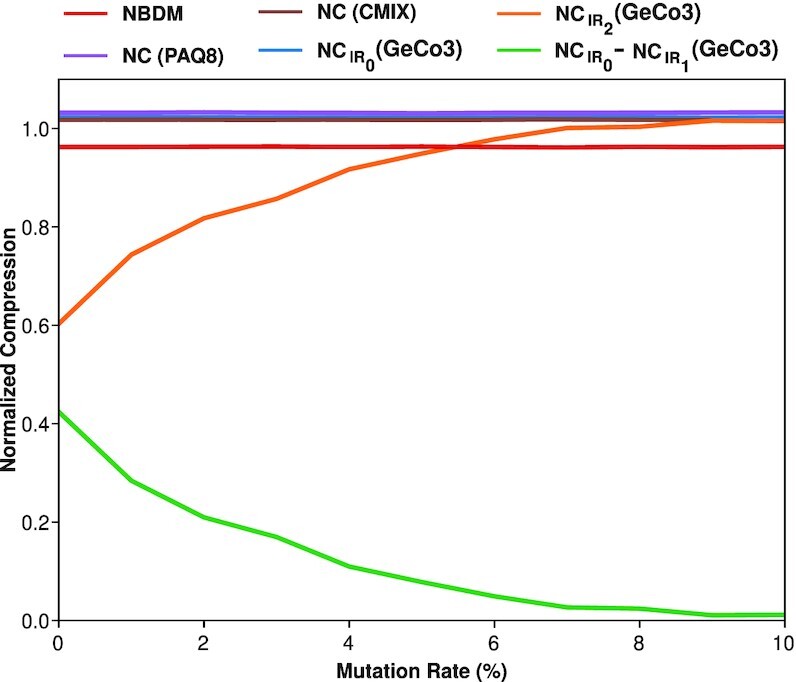
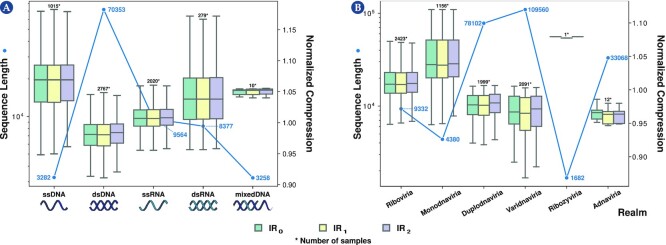
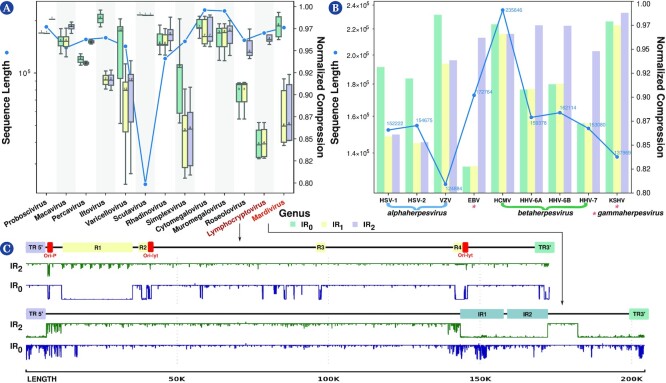
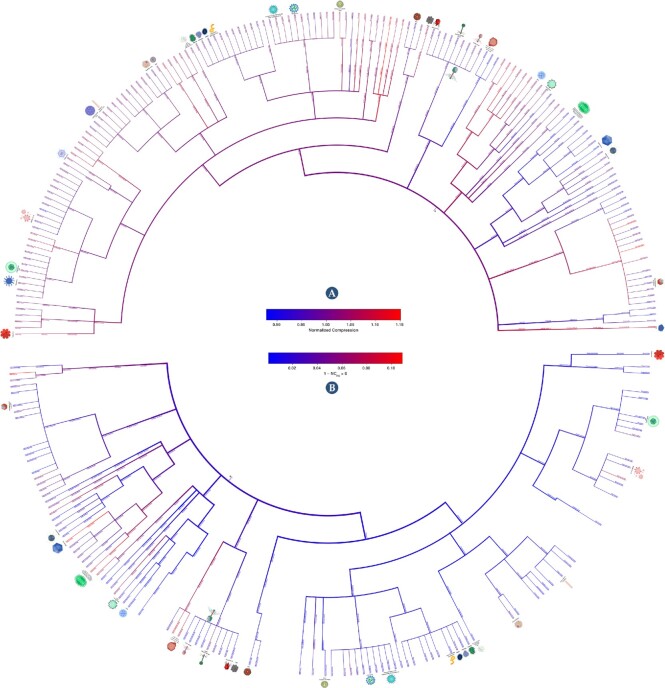
This work provides a comprehensive landscape of the viral genome's complexity (or quantity of information), identifying the most redundant and complex groups regarding their genome sequence while providing their distribution and characteristics at a large and local scale. Moreover, we identify and quantify inverted repeats abundance in viral genomes. For this purpose, we measure the sequence complexity of each available viral genome using data compression, demonstrating that adequate data compressors can efficiently quantify the complexity of viral genome sequences, including subsequences better represented by algorithmic sources (e.g., inverted repeats). Using a state-of-the-art genomic compressor on an extensive viral genomes database, we show that double-stranded DNA viruses are, on average, the most redundant viruses while single-stranded DNA viruses are the least. Contrarily, double-stranded RNA viruses show a lower redundancy relative to single-stranded RNA. Furthermore, we extend the ability of data compressors to quantify local complexity (or information content) in viral genomes using complexity profiles, unprecedently providing a direct complexity analysis of human herpesviruses. We also conceive a features-based classification methodology that can accurately distinguish viral genomes at different taxonomic levels without direct comparisons between sequences. This methodology combines data compression with simple measures such as GC-content percentage and sequence length, followed by machine learning classifiers.
This article presents methodologies and findings that are highly relevant for understanding the patterns of similarity and singularity between viral groups, opening new frontiers for studying viral genomes' organization while depicting the complexity trends and classification components of these genomes at different taxonomic levels. The whole study is supported by an extensive website (https://asilab.github.io/canvas/) for comprehending the viral genome characterization using dynamic and interactive approaches.
病毒是拥有最小感染细胞、适应、繁殖和生存指令的最短但高度丰富的物种之一。然而,随着当前大量病毒基因组序列的可用性,科学界缺乏一种自动揭示病毒基因组组织、关系和基本特征的复杂性景观。
这项工作提供了病毒基因组复杂性(或信息量)的综合景观,确定了关于其基因组序列最冗余和最复杂的组,并提供了它们在大尺度和局部尺度上的分布和特征。此外,我们识别和量化了病毒基因组中反转重复的丰度。为此,我们使用数据压缩测量每个可用病毒基因组的序列复杂性,证明了适当的数据压缩器可以有效地量化病毒基因组序列的复杂性,包括算法源(例如反转重复)更好表示的子序列。我们在广泛的病毒基因组数据库上使用最先进的基因组压缩器,表明双链 DNA 病毒平均是最冗余的病毒,而单链 DNA 病毒是最少的。相反,双链 RNA 病毒相对于单链 RNA 显示出较低的冗余性。此外,我们扩展了数据压缩器在病毒基因组中量化局部复杂性(或信息量)的能力,使用复杂性剖面,前所未有地提供了人类疱疹病毒的直接复杂性分析。我们还设想了一种基于特征的分类方法,该方法可以在不进行序列直接比较的情况下,准确地区分不同分类水平的病毒基因组。该方法结合了数据压缩和简单的措施,如 GC 含量百分比和序列长度,然后是机器学习分类器。
本文提出的方法和发现对于理解病毒群体之间的相似性和独特性模式具有重要意义,为研究病毒基因组的组织开辟了新的前沿,同时描绘了这些基因组在不同分类水平上的复杂性趋势和分类组成。整个研究得到了一个广泛的网站(https://asilab.github.io/canvas/)的支持,用于使用动态和交互式方法理解病毒基因组的特征。