Rajkhowa Swaraj, Scaria Joy, Garcia Daniel L, Musser Kimberlee A, Akey Bruce L, Chang Yung-Fu
Department of Population Medicine and Diagnostic Sciences, College of Veterinary Medicine, Cornell University, Ithaca, New York 14853, USA.
BMC Res Notes. 2010 Dec 21;3:343. doi: 10.1186/1756-0500-3-343.
Although many strain typing methods exist for pathogenic Escherichia coli, most have drawbacks in terms of resolving power, interpretability, or scalability. For this reason, multilocus sequence typing (MLST) is an appealing alternative especially when applied to the typing of temporal and spatially separated isolates. This method relies on an unambiguous DNA sequence analysis of nucleotide polymorphisms in housekeeping genes and has shown a high degree of intraspecies discriminatory power for bacterial and fungal pathogens.
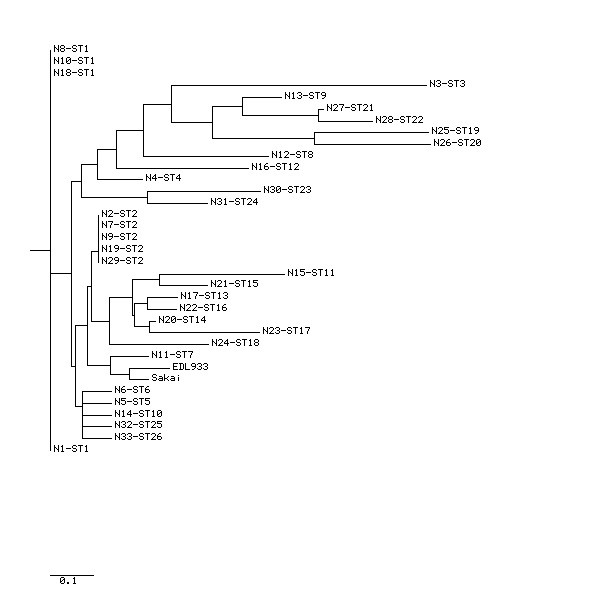
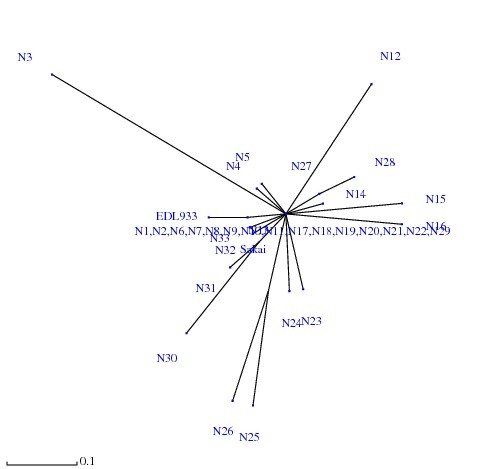
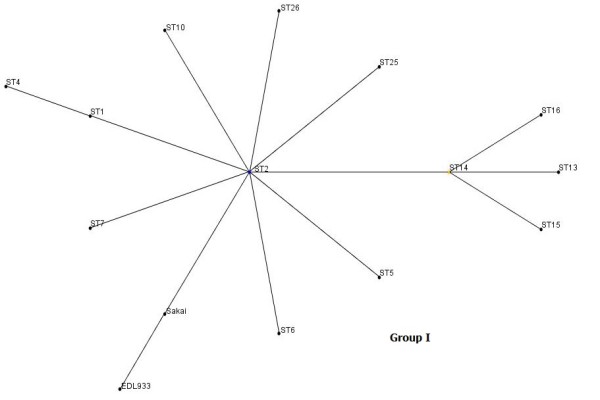
Here we used the MLST method to study the genetic diversity among E. coli O157 isolates collected from humans from two different locations of USA over a period of several years (2000-2008). MLST analysis of 33 E. coli O157 patient isolates using the eBurst algorithm distinguished 26 different sequence types (STs), which were clustered into two clonal groups and 11 singletons. The predominant ST was ST2, which consisted of 5 isolates (14.28%) followed by ST1 (11.42%). All the isolates under clonal group I exhibited a virtually similar virulence profile except for two strains, which tested negative for the presence of stx genes. The isolates that were assigned to clonal group II in addition to the 11 singletons were found to be phylogenetically distant from clonal group I. Furthermore, we observed a positive correlation between the virulence profile of the isolates and their clonal origin.
Our data suggests the presence of genetic diversity among E. coli O157 isolates from humans shows no measurable correlation to the geographic origin of the isolates.
尽管存在多种用于致病性大肠杆菌的菌株分型方法,但大多数在分辨能力、可解释性或可扩展性方面存在缺陷。因此,多位点序列分型(MLST)是一种有吸引力的替代方法,尤其适用于对在时间和空间上分离的菌株进行分型。该方法依赖于对管家基因中核苷酸多态性进行明确的DNA序列分析,并且已显示出对细菌和真菌病原体具有高度的种内鉴别能力。
在这里,我们使用MLST方法研究了在数年(2000 - 2008年)期间从美国两个不同地点的人类中分离出的大肠杆菌O157菌株之间的遗传多样性。使用eBurst算法对33株大肠杆菌O157患者分离株进行MLST分析,区分出26种不同的序列类型(STs),这些序列类型被聚类为两个克隆组和11个单株。主要的ST是ST2,由5株分离株组成(14.28%),其次是ST1(11.42%)。除了两株stx基因检测呈阴性的菌株外,克隆组I中的所有分离株都表现出几乎相似的毒力谱。除了11个单株外,被分配到克隆组II的分离株在系统发育上与克隆组I距离较远。此外,我们观察到分离株的毒力谱与其克隆起源之间存在正相关。
我们的数据表明,来自人类的大肠杆菌O157分离株之间存在遗传多样性,且与分离株的地理起源没有可测量的相关性。