Biology Department, City University of New York, Queens College, 65-30 Kissena Blvd, Flushing, NY 11367-1597, USA.
BMC Bioinformatics. 2011 Jan 3;12:1. doi: 10.1186/1471-2105-12-1.
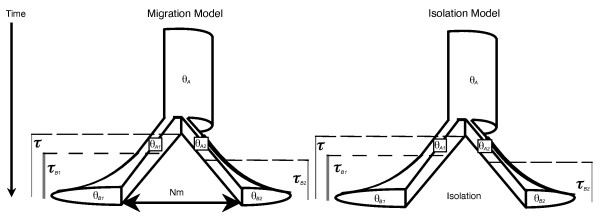
MTML-msBayes uses hierarchical approximate Bayesian computation (HABC) under a coalescent model to infer temporal patterns of divergence and gene flow across codistributed taxon-pairs. Under a model of multiple codistributed taxa that diverge into taxon-pairs with subsequent gene flow or isolation, one can estimate hyper-parameters that quantify the mean and variability in divergence times or test models of migration and isolation. The software uses multi-locus DNA sequence data collected from multiple taxon-pairs and allows variation across taxa in demographic parameters as well as heterogeneity in DNA mutation rates across loci. The method also allows a flexible sampling scheme: different numbers of loci of varying length can be sampled from different taxon-pairs.
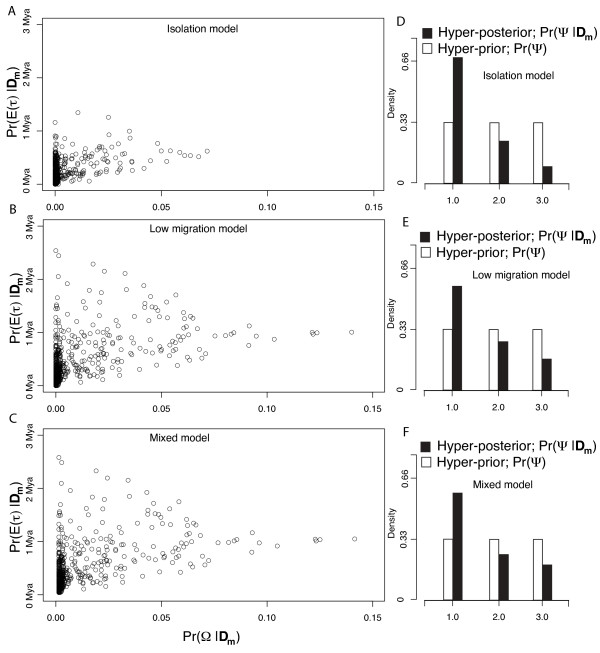
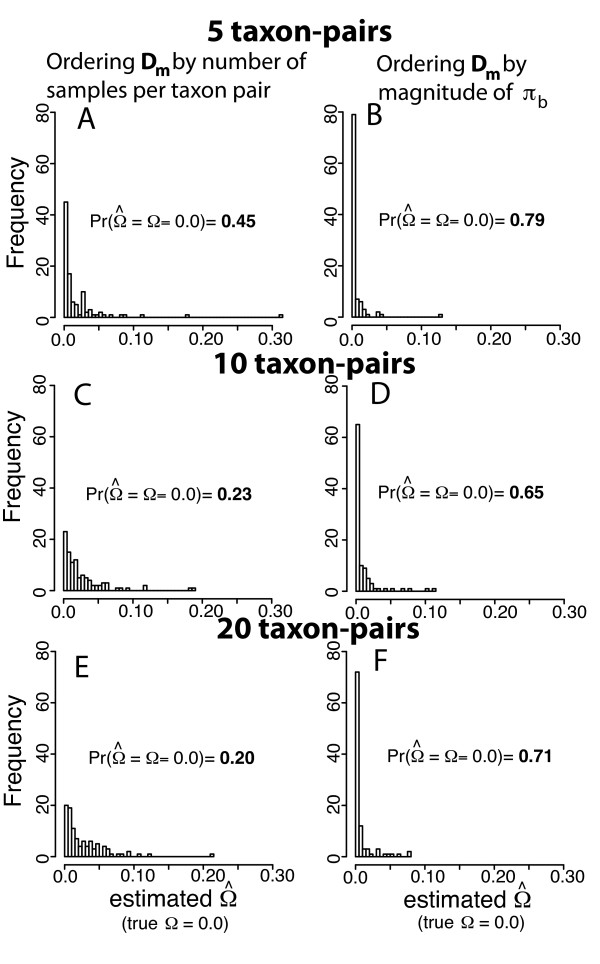
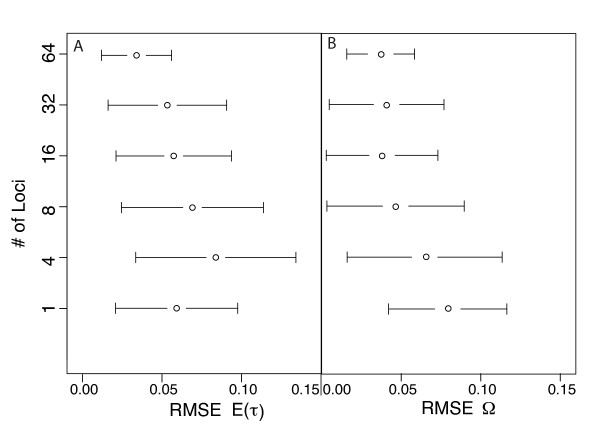
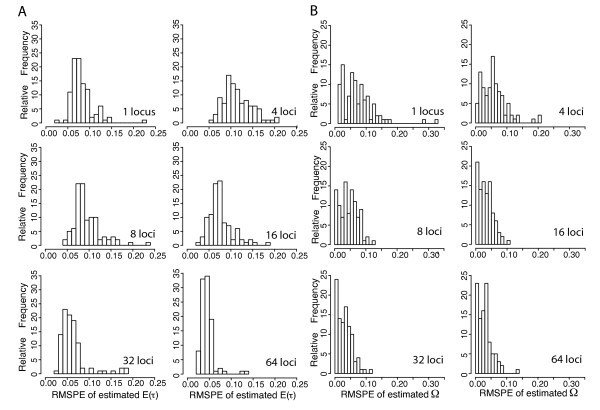
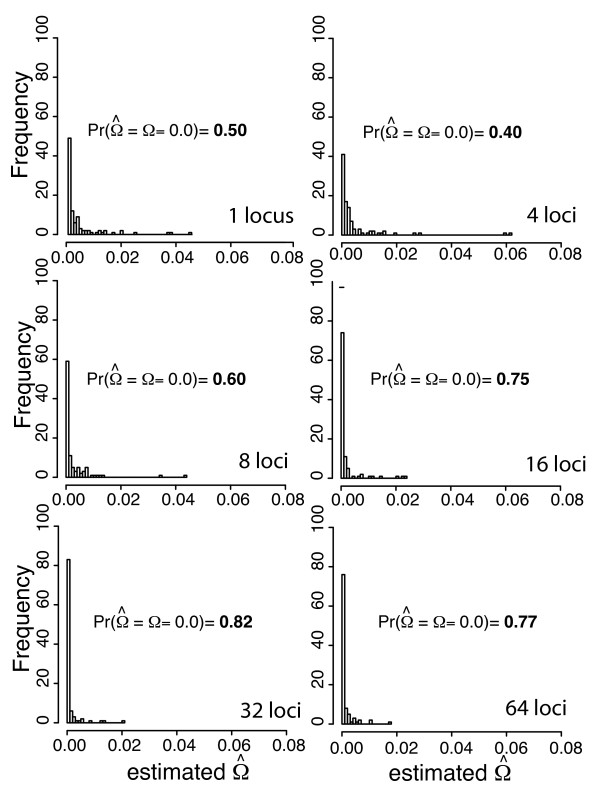
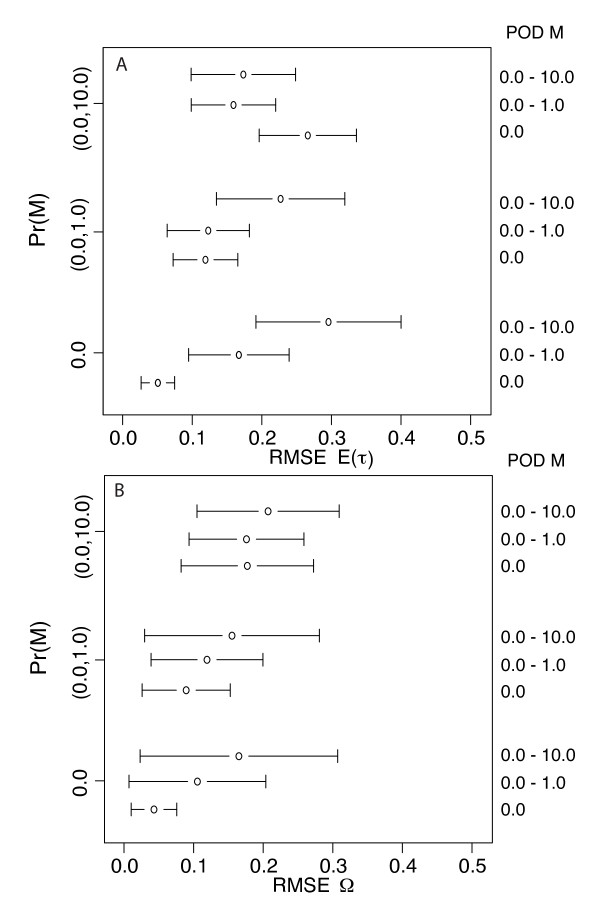
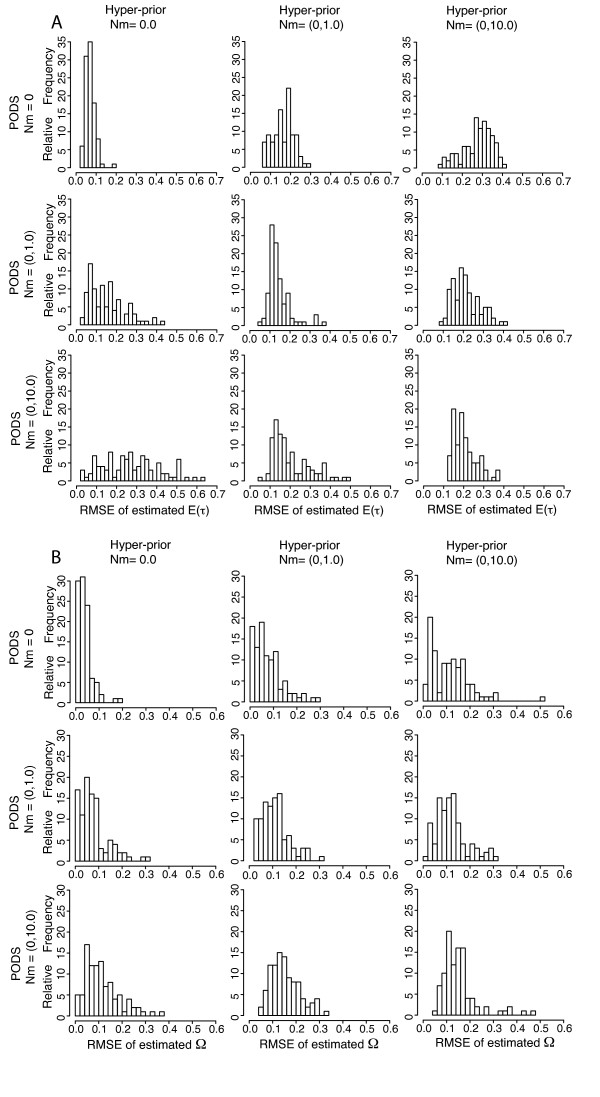
Simulation tests reveal increasing power with increasing numbers of loci when attempting to distinguish temporal congruence from incongruence in divergence times across taxon-pairs. These results are robust to DNA mutation rate heterogeneity. Estimating mean divergence times and testing simultaneous divergence was less accurate with migration, but improved if one specified the correct migration model. Simulation validation tests demonstrated that one can detect the correct migration or isolation model with high probability, and that this HABC model testing procedure was greatly improved by incorporating a summary statistic originally developed for this task (Wakeley's ΨW). The method is applied to an empirical data set of three Australian avian taxon-pairs and a result of simultaneous divergence with some subsequent gene flow is inferred.
To retain flexibility and compatibility with existing bioinformatics tools, MTML-msBayes is a pipeline software package consisting of Perl, C and R programs that are executed via the command line. Source code and binaries are available for download at http://msbayes.sourceforge.net/ under an open source license (GNU Public License).
MTML-msBayes 使用分支近似贝叶斯计算(HABC)在一个融合模型下推断跨越共分布分类单元对的分歧和基因流动的时间模式。在一个多个共分布分类单元分裂成具有随后基因流动或隔离的分类单元对的模型下,人们可以估计量化分歧时间均值和变异性的超参数,或者测试迁移和隔离模型。该软件使用从多个分类单元对收集的多基因座 DNA 序列数据,并允许在人口参数方面跨越分类单元的变化,以及在跨基因座的 DNA 突变率方面的异质性。该方法还允许灵活的抽样方案:可以从不同的分类单元对中抽样不同数量的不同长度的基因座。
模拟测试表明,当试图区分分类单元对之间分歧时间的一致性和不一致性时,随着基因座数量的增加,其能力逐渐增强。这些结果对于 DNA 突变率异质性是稳健的。估计平均分歧时间和测试同时分歧的准确性较低,如果指定了正确的迁移模型则会提高。模拟验证测试表明,人们可以以很高的概率检测到正确的迁移或隔离模型,并且通过结合最初为此任务开发的汇总统计信息(Wakeley 的 ΨW),这种 HABC 模型测试程序得到了极大的改进。该方法应用于三个澳大利亚鸟类分类单元对的一个经验数据集,推断出同时分歧且有一些后续基因流动的结果。
为了保持灵活性和与现有生物信息学工具的兼容性,MTML-msBayes 是一个由 Perl、C 和 R 程序组成的管道软件包,通过命令行执行。源代码和二进制文件可在 http://msbayes.sourceforge.net/ 上下载,采用开源许可证(GNU 公共许可证)。