Cheng Tammy M K, Lu Yu-En, Vendruscolo Michele, Lio' Pietro, Blundell Tom L
Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom.
PLoS Comput Biol. 2008 Jul 25;4(7):e1000135. doi: 10.1371/journal.pcbi.1000135.
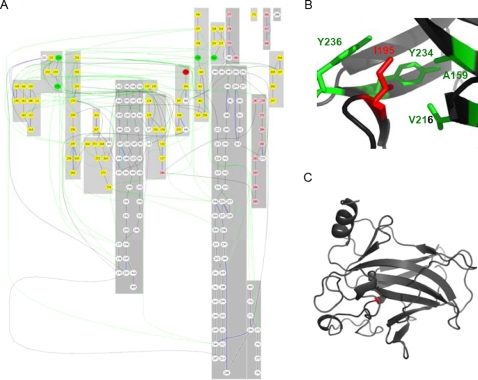
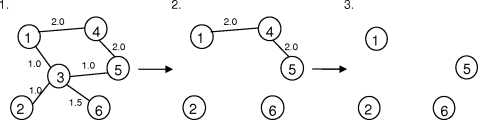
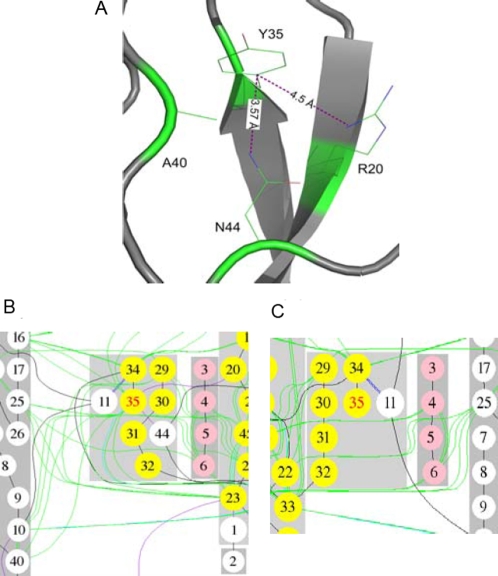
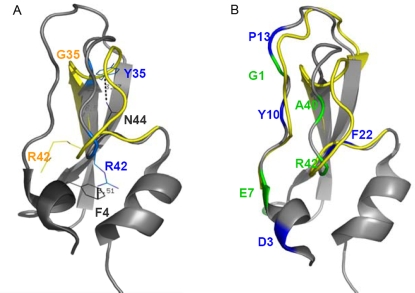
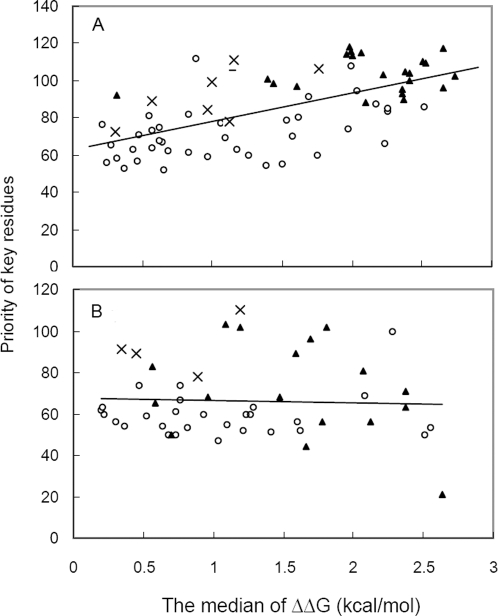
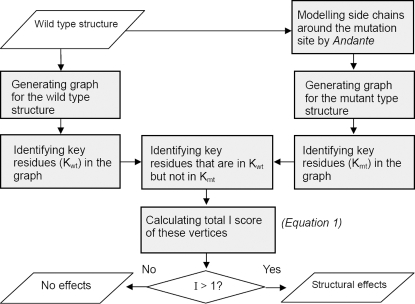
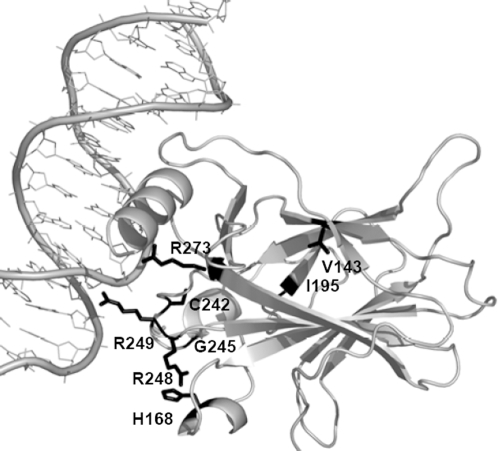
Recent analyses of human genome sequences have given rise to impressive advances in identifying non-synonymous single nucleotide polymorphisms (nsSNPs). By contrast, the annotation of nsSNPs and their links to diseases are progressing at a much slower pace. Many of the current approaches to analysing disease-associated nsSNPs use primarily sequence and evolutionary information, while structural information is relatively less exploited. In order to explore the potential of such information, we developed a structure-based approach, Bongo (Bonds ON Graph), to predict structural effects of nsSNPs. Bongo considers protein structures as residue-residue interaction networks and applies graph theoretical measures to identify the residues that are critical for maintaining structural stability by assessing the consequences on the interaction network of single point mutations. Our results show that Bongo is able to identify mutations that cause both local and global structural effects, with a remarkably low false positive rate. Application of the Bongo method to the prediction of 506 disease-associated nsSNPs resulted in a performance (positive predictive value, PPV, 78.5%) similar to that of PolyPhen (PPV, 77.2%) and PANTHER (PPV, 72.2%). As the Bongo method is solely structure-based, our results indicate that the structural changes resulting from nsSNPs are closely associated to their pathological consequences.
近期对人类基因组序列的分析在识别非同义单核苷酸多态性(nsSNPs)方面取得了令人瞩目的进展。相比之下,nsSNPs的注释及其与疾病的关联进展要缓慢得多。当前许多分析与疾病相关的nsSNPs的方法主要利用序列和进化信息,而结构信息的利用相对较少。为了探索此类信息的潜力,我们开发了一种基于结构的方法Bongo(图上的键)来预测nsSNPs的结构效应。Bongo将蛋白质结构视为残基-残基相互作用网络,并应用图论方法通过评估单点突变对相互作用网络的影响来识别对维持结构稳定性至关重要的残基。我们的结果表明,Bongo能够识别出引起局部和全局结构效应的突变,假阳性率极低。将Bongo方法应用于预测506个与疾病相关的nsSNPs,其性能(阳性预测值,PPV,78.5%)与PolyPhen(PPV,77.2%)和PANTHER(PPV,72.2%)相似。由于Bongo方法完全基于结构,我们的结果表明,nsSNPs导致的结构变化与其病理后果密切相关。