Laboratory of Pharmacology, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA.
J Neuroinflammation. 2011 Jan 13;8(1):3. doi: 10.1186/1742-2094-8-3.
β-Amyloid peptide (Aβ) is a major protein in the brain associated with Alzheimer's and Parkinson's diseases. The purpose of this study was to investigate the role of macrophage antigen-1 (MAC1) receptor, an integrin scavenger receptor in microglia, and subsequent signaling events in mediating Aβ-induced neurotoxicity. We have previously reported that NADPH oxidase (PHOX) on microglia and superoxide produced by PHOX are critical for Aβ-induced loss of dopaminergic neurons. However, the upstream signaling pathway of superoxide production remains unclear.
For the in vitro study, mesencephalic neuron-glia cultures and microglia-enriched cultures from mice deficient in the MAC1 receptor (MAC1-/-) and wild type controls were used to investigate the role of MAC1 receptor in Aβ-induced neurotoxicity and the role of phosphoinositide-3 kinase (PI3K) in the signal pathway between MAC1 receptor and PHOX. For the in vivo study, Aβ was injected into the substantia nigra of MAC1-/- mice and wild type mice to confirm the role of MAC1 receptor.
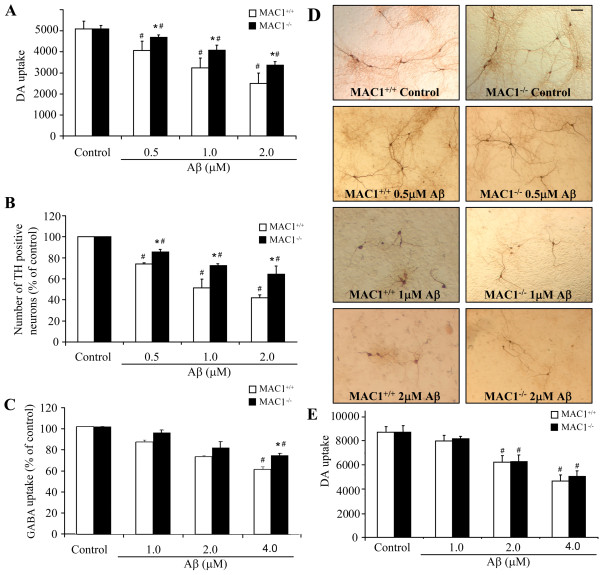
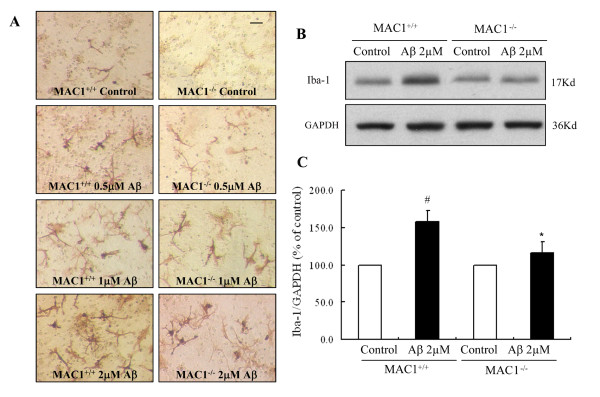
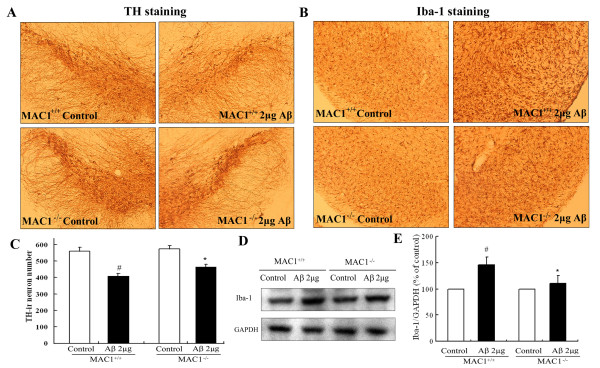
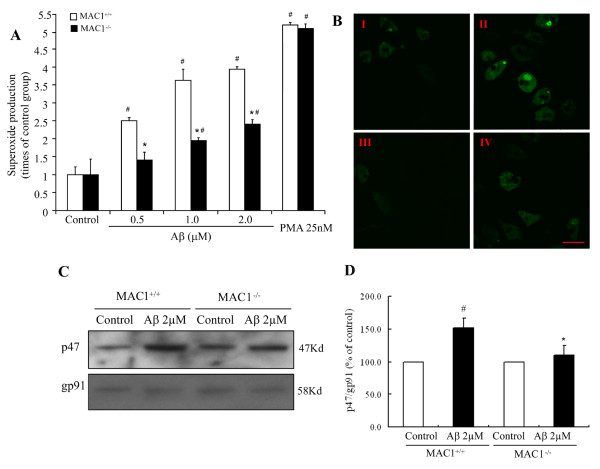
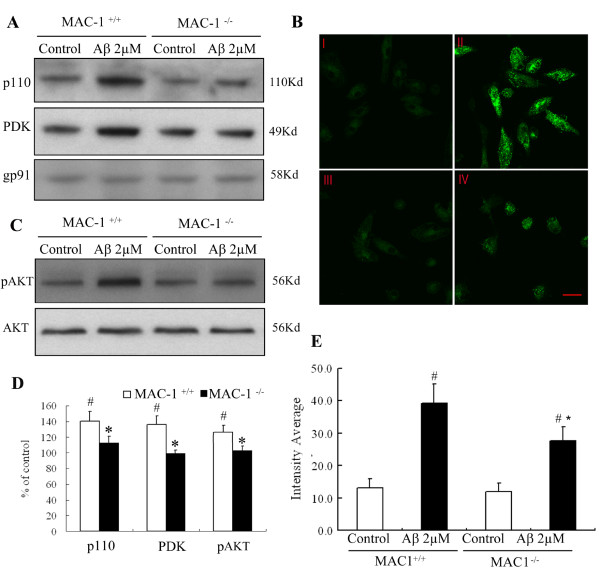
We found that Aβ-induced activation of microglia, activation of PHOX, generation of superoxide and other reactive oxygen species, and loss of dopaminergic neurons were decreased in MAC1-/- cultures compared to MAC1+/+ cultures. In MAC1-/- mice, dopaminergic neuron loss in response to Aβ injection into the substantia nigra was reduced relative to MAC1+/+ mice. Thus, MAC1 receptor-mediated PHOX activation and increased superoxide production are associated with Aβ-induced neurotoxicity. PI3K activation was one downstream step in MAC1 signaling to PHOX and played an important role in Aβ-induced neurotoxicity. In microglia-enriched cultures from MAC1-/- mice, Aβ-induced activation of PI3K (phosphorylation of target proteins and PIP3 production) was reduced relative to MAC1+/+ cultures.
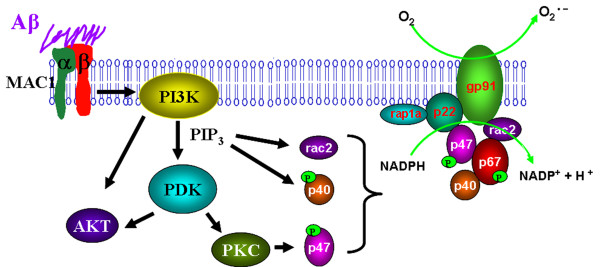
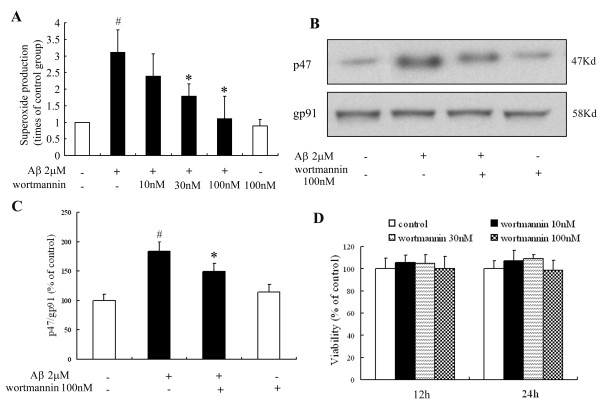
Taken together, our data demonstrate that Aβ activates MAC1 receptor to increase the activity of PI3K, which in turn phosphorylates p47phox, triggers the translocation of cytosolic subunits of PHOX to microglia membrane, increases PHOX activation and the subsequent production of superoxide and causes neurotoxicity.
β-淀粉样肽(Aβ)是大脑中与阿尔茨海默病和帕金森病相关的主要蛋白质。本研究的目的是探讨巨噬细胞抗原-1(MAC1)受体(小胶质细胞中的整合素清道夫受体)的作用,以及随后的信号事件在介导 Aβ 诱导的神经毒性中的作用。我们之前报道过,小胶质细胞中的 NADPH 氧化酶(PHOX)和 PHOX 产生的超氧阴离子对于 Aβ 诱导的多巴胺能神经元丧失至关重要。然而,超氧阴离子产生的上游信号通路仍不清楚。
在体外研究中,使用缺乏 MAC1 受体(MAC1-/-)的小鼠的中脑神经元-胶质细胞培养物和富含小胶质细胞的培养物以及野生型对照,来研究 MAC1 受体在 Aβ 诱导的神经毒性中的作用以及磷酸肌醇 3 激酶(PI3K)在 MAC1 受体和 PHOX 之间信号通路中的作用。在体内研究中,将 Aβ 注射到 MAC1-/-小鼠和野生型小鼠的黑质中,以确认 MAC1 受体的作用。
我们发现,与 MAC1+/+培养物相比,Aβ 诱导的小胶质细胞活化、PHOX 活化、超氧阴离子和其他活性氧的产生以及多巴胺能神经元的丧失在 MAC1-/-培养物中减少。在 MAC1-/-小鼠中,与 MAC1+/+小鼠相比,Aβ 注射到黑质中引起的多巴胺能神经元丧失减少。因此,MAC1 受体介导的 PHOX 活化和超氧阴离子产生的增加与 Aβ 诱导的神经毒性有关。PI3K 激活是 MAC1 信号转导至 PHOX 的下游步骤之一,在 Aβ 诱导的神经毒性中发挥重要作用。在 MAC1-/-小鼠的富含小胶质细胞的培养物中,与 MAC1+/+培养物相比,Aβ 诱导的 PI3K 活化(靶蛋白的磷酸化和 PIP3 的产生)减少。
综上所述,我们的数据表明,Aβ 激活 MAC1 受体以增加 PI3K 的活性,PI3K 进而磷酸化 p47phox,触发 PHOX 胞质亚基向小胶质细胞膜的易位,增加 PHOX 的活化和随后的超氧阴离子的产生,并导致神经毒性。