Department of Bioinformatics, Institute of Microbiology and Genetics, Georg-August-University Göttingen, Göttingen, Germany.
BMC Genomics. 2009 Nov 12;10:520. doi: 10.1186/1471-2164-10-520.
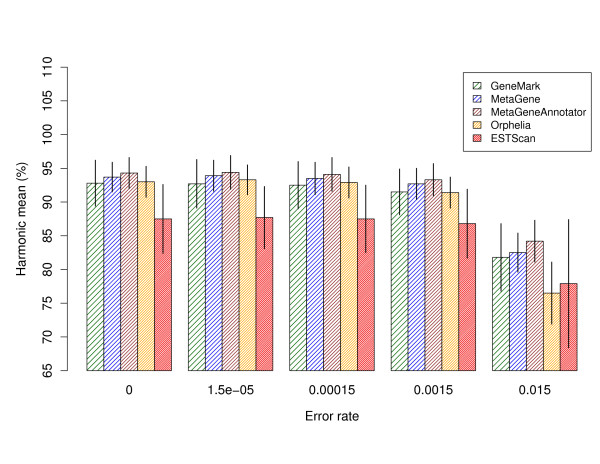
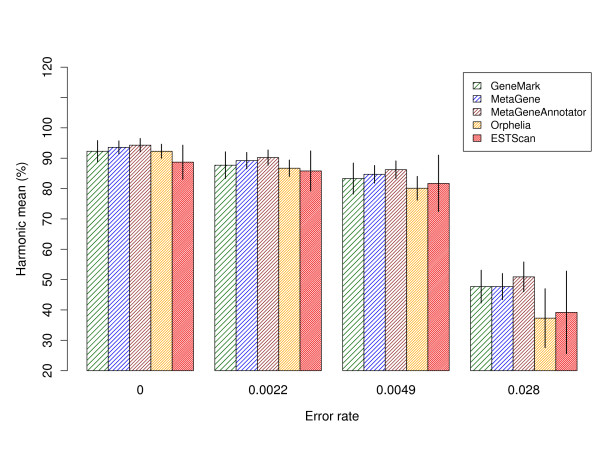
Gene prediction is an essential step in the annotation of metagenomic sequencing reads. Since most metagenomic reads cannot be assembled into long contigs, specialized statistical gene prediction tools have been developed for short and anonymous DNA fragments, e.g. MetaGeneAnnotator and Orphelia. While conventional gene prediction methods have been subject to a benchmark study on real sequencing reads with typical errors, such a comparison has not been conducted for specialized tools, yet. Their gene prediction accuracy was mostly measured on error free DNA fragments.
In this study, Sanger and pyrosequencing reads were simulated on the basis of models that take all types of sequencing errors into account. All metagenomic gene prediction tools showed decreasing accuracy with increasing sequencing error rates. Performance results on an established metagenomic benchmark dataset are also reported. In addition, we demonstrate that ESTScan, a tool for sequencing error compensation in eukaryotic expressed sequence tags, outperforms some metagenomic gene prediction tools on reads with high error rates although it was not designed for the task at hand.
This study fills an important gap in metagenomic gene prediction research. Specialized methods are evaluated and compared with respect to sequencing error robustness. Results indicate that the integration of error-compensating methods into metagenomic gene prediction tools would be beneficial to improve metagenome annotation quality.
基因预测是对宏基因组测序reads 进行注释的重要步骤。由于大多数宏基因组reads 无法组装成长的连续序列,因此专门开发了统计基因预测工具来处理短的、匿名的 DNA 片段,例如 MetaGeneAnnotator 和 Orphelia。虽然传统的基因预测方法已经在具有典型错误的真实测序reads 上进行了基准测试,但尚未对专门的工具进行此类比较。它们的基因预测准确性主要是在没有错误的 DNA 片段上进行测量的。
在这项研究中,根据考虑了所有类型测序错误的模型,对 Sanger 和焦磷酸测序reads 进行了模拟。所有宏基因组基因预测工具的准确性都随着测序错误率的增加而降低。还报告了在已建立的宏基因组基准数据集上的性能结果。此外,我们证明了 ESTScan,一种用于补偿真核表达序列标签中测序错误的工具,尽管它不是为此任务设计的,但在高错误率的reads 上优于某些宏基因组基因预测工具。
这项研究填补了宏基因组基因预测研究中的一个重要空白。专门的方法针对测序错误稳健性进行了评估和比较。结果表明,将纠错方法集成到宏基因组基因预测工具中有助于提高宏基因组注释的质量。