Hackenberg Michael, Carpena Pedro, Bernaola-Galván Pedro, Barturen Guillermo, Alganza Angel M, Oliver José L
Dpto, de Genética, Facultad de Ciencias, Universidad de Granada, Campus de Fuentenueva s/n, 18071-Granada & Lab, de Bioinformática, Centro de Investigación Biomédica, PTS, Avda, del Conocimiento s/n, 18100-Granada, Spain.
Algorithms Mol Biol. 2011 Jan 24;6:2. doi: 10.1186/1748-7188-6-2.
Many k-mers (or DNA words) and genomic elements are known to be spatially clustered in the genome. Well established examples are the genes, TFBSs, CpG dinucleotides, microRNA genes and ultra-conserved non-coding regions. Currently, no algorithm exists to find these clusters in a statistically comprehensible way. The detection of clustering often relies on densities and sliding-window approaches or arbitrarily chosen distance thresholds.
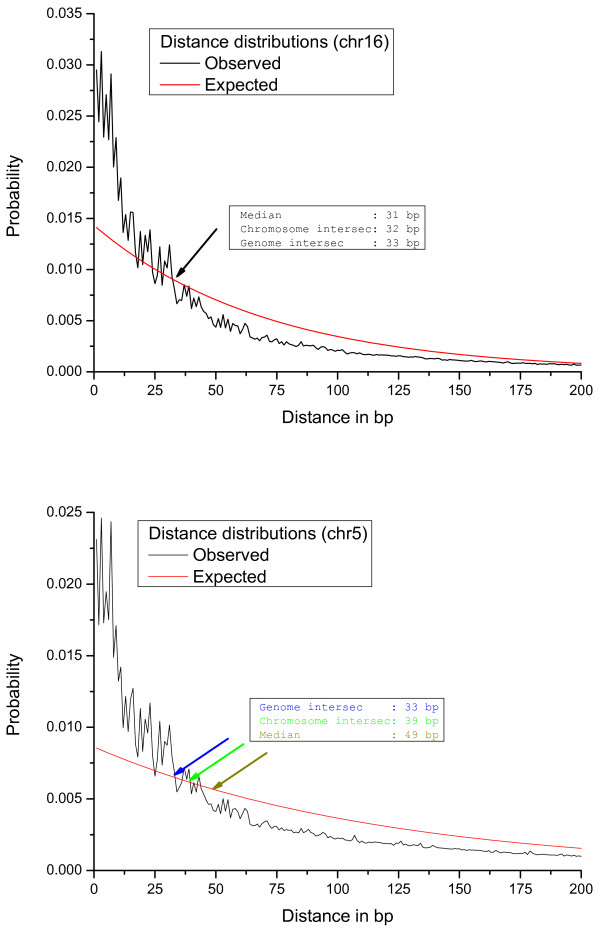
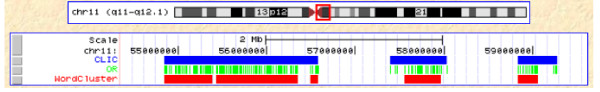
We introduce here an algorithm to detect clusters of DNA words (k-mers), or any other genomic element, based on the distance between consecutive copies and an assigned statistical significance. We implemented the method into a web server connected to a MySQL backend, which also determines the co-localization with gene annotations. We demonstrate the usefulness of this approach by detecting the clusters of CAG/CTG (cytosine contexts that can be methylated in undifferentiated cells), showing that the degree of methylation vary drastically between inside and outside of the clusters. As another example, we used WordCluster to search for statistically significant clusters of olfactory receptor (OR) genes in the human genome.
WordCluster seems to predict biological meaningful clusters of DNA words (k-mers) and genomic entities. The implementation of the method into a web server is available at http://bioinfo2.ugr.es/wordCluster/wordCluster.php including additional features like the detection of co-localization with gene regions or the annotation enrichment tool for functional analysis of overlapped genes.
已知许多k-mer(或DNA单词)和基因组元件在基因组中呈空间聚集状态。已明确的例子包括基因、转录因子结合位点(TFBS)、CpG二核苷酸、微小RNA基因和超保守非编码区。目前,尚无一种算法能够以统计学上可理解的方式找到这些聚集区域。聚集区域的检测通常依赖于密度和滑动窗口方法或任意选择的距离阈值。
我们在此介绍一种基于连续拷贝之间的距离和指定的统计显著性来检测DNA单词(k-mer)或任何其他基因组元件聚集区域的算法。我们将该方法实现为一个连接到MySQL后端的网络服务器,该服务器还能确定与基因注释的共定位情况。我们通过检测CAG/CTG(在未分化细胞中可甲基化的胞嘧啶环境)的聚集区域,证明了这种方法的有效性,结果表明聚集区域内外的甲基化程度差异很大。作为另一个例子,我们使用WordCluster在人类基因组中搜索嗅觉受体(OR)基因的具有统计学显著性的聚集区域。
WordCluster似乎能够预测DNA单词(k-mer)和基因组实体的具有生物学意义的聚集区域。该方法在网络服务器上的实现可通过http://bioinfo2.ugr.es/wordCluster/wordCluster.php获取,其中还包括与基因区域共定位检测或重叠基因功能分析的注释富集工具等其他功能。