Machado Moara, Magalhães Wagner Cs, Sene Allan, Araújo Bruno, Faria-Campos Alessandra C, Chanock Stephen J, Scott Leandro, Oliveira Guilherme, Tarazona-Santos Eduardo, Rodrigues Maira R
Departamento de Biologia Geral, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av Antonio Carlos 6627, Pampulha, Caixa Postal 486, Belo Horizonte, MG, CEP 31270-910, Brazil.
Departamento de Ciência da Computação, Instituto de Ciências Exatas, Universidade Federal de Minas Gerais, Av Antonio Carlos 6627, Pampulha, Belo Horizonte, MG, CEP 31270-910, Brazil.
Investig Genet. 2011 Feb 1;2(1):3. doi: 10.1186/2041-2223-2-3.
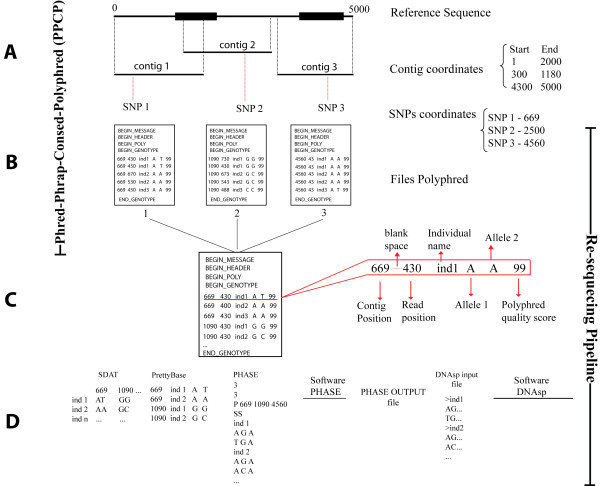
Targeted re-sequencing is one of the most powerful and widely used strategies for population genetics studies because it allows an unbiased screening for variation that is suitable for a wide variety of organisms. Examples of studies that require re-sequencing data are evolutionary inferences, epidemiological studies designed to capture rare polymorphisms responsible for complex traits and screenings for mutations in families and small populations with high incidences of specific genetic diseases. Despite the advent of next-generation sequencing technologies, Sanger sequencing is still the most popular approach in population genetics studies because of the widespread availability of automatic sequencers based on capillary electrophoresis and because it is still less prone to sequencing errors, which is critical in population genetics studies. Two popular software applications for re-sequencing studies are Phred-Phrap-Consed-Polyphred, which performs base calling, alignment, graphical edition and genotype calling and DNAsp, which performs a set of population genetics analyses. These independent tools are the start and end points of basic analyses. In between the use of these tools, there is a set of basic but error-prone tasks to be performed with re-sequencing data.
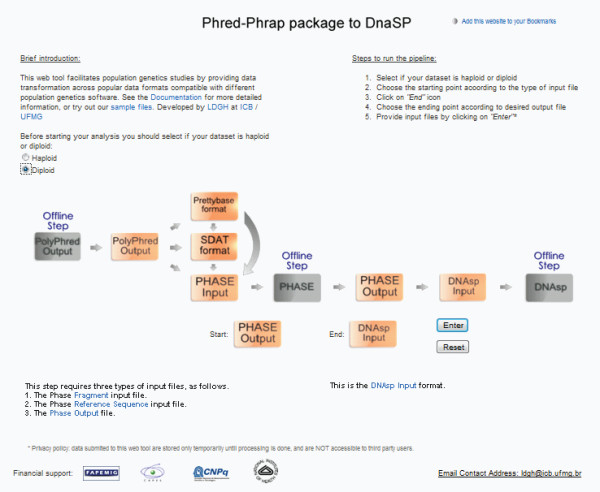
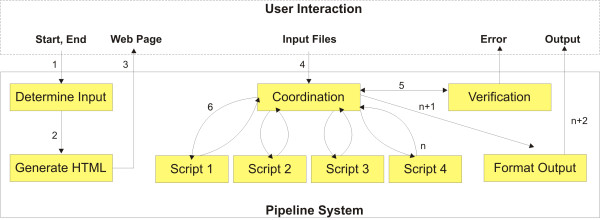
In order to assist with these intermediate tasks, we developed a pipeline that facilitates data handling typical of re-sequencing studies. Our pipeline: (1) consolidates different outputs produced by distinct Phred-Phrap-Consed contigs sharing a reference sequence; (2) checks for genotyping inconsistencies; (3) reformats genotyping data produced by Polyphred into a matrix of genotypes with individuals as rows and segregating sites as columns; (4) prepares input files for haplotype inferences using the popular software PHASE; and (5) handles PHASE output files that contain only polymorphic sites to reconstruct the inferred haplotypes including polymorphic and monomorphic sites as required by population genetics software for re-sequencing data such as DNAsp.
We tested the pipeline in re-sequencing studies of haploid and diploid data in humans, plants, animals and microorganisms and observed that it allowed a substantial decrease in the time required for sequencing analyses, as well as being a more controlled process that eliminates several classes of error that may occur when handling datasets. The pipeline is also useful for investigators using other tools for sequencing and population genetics analyses.
靶向重测序是群体遗传学研究中最强大且应用最广泛的策略之一,因为它能够对变异进行无偏筛选,适用于多种生物。需要重测序数据的研究实例包括进化推断、旨在捕获负责复杂性状的罕见多态性的流行病学研究,以及对特定遗传疾病高发的家庭和小群体中的突变进行筛查。尽管新一代测序技术已经出现,但由于基于毛细管电泳的自动测序仪广泛可用,且在群体遗传学研究中仍然不太容易出现测序错误,桑格测序仍然是群体遗传学研究中最受欢迎的方法。两种用于重测序研究的流行软件应用程序是Phred-Phrap-Consed-Polyphred,它执行碱基识别、比对、图形编辑和基因型识别,以及DNAsp,它执行一组群体遗传学分析。这些独立的工具是基础分析的起点和终点。在使用这些工具的过程中,对于重测序数据有一组基本但容易出错的任务需要执行。
为了协助完成这些中间任务,我们开发了一个流程,便于处理重测序研究中典型的数据。我们的流程:(1)整合由共享参考序列的不同Phred-Phrap-Consed重叠群产生的不同输出;(2)检查基因分型的不一致性;(3)将Polyphred产生的基因分型数据重新格式化为一个基因型矩阵,其中个体为行,分离位点为列;(4)使用流行软件PHASE准备用于单倍型推断的输入文件;(5)处理仅包含多态性位点的PHASE输出文件,以根据群体遗传学软件(如DNAsp)对重测序数据的要求,重建包括多态性和单态性位点的推断单倍型。
我们在人类、植物、动物和微生物的单倍体和二倍体数据的重测序研究中测试了该流程,观察到它显著减少了测序分析所需的时间,并且是一个更可控的过程,消除了处理数据集时可能出现的几类错误。该流程对于使用其他测序和群体遗传学分析工具的研究人员也很有用。