Division of Cardiology, Department of Medicine, Yonsei University College of Medicine, Seoul, Korea.
Yonsei Med J. 2011 Mar;52(2):211-9. doi: 10.3349/ymj.2011.52.2.211.
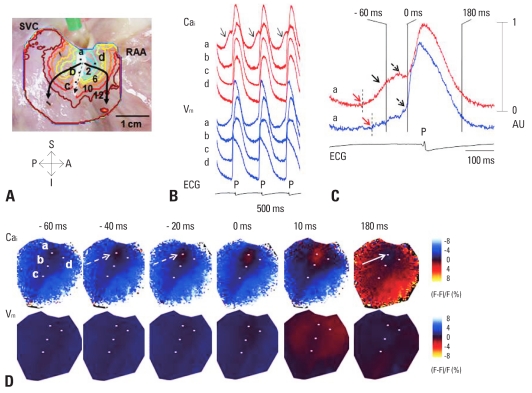
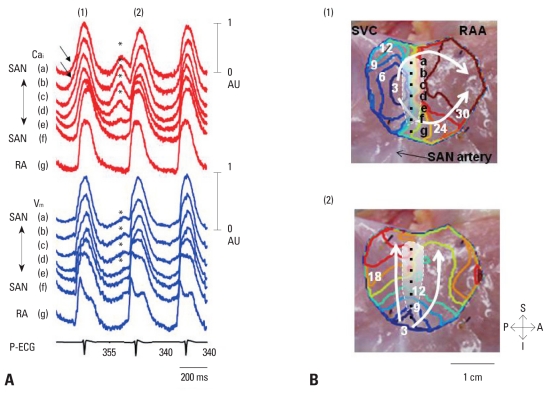
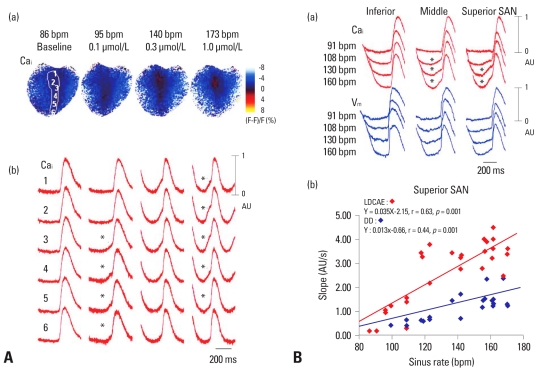
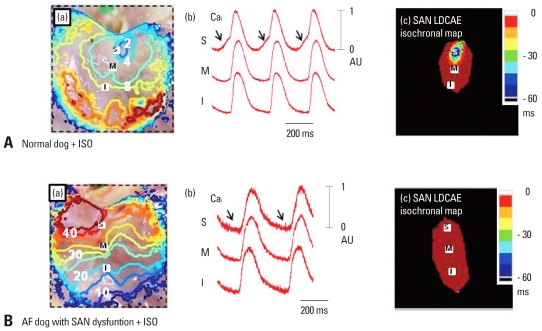
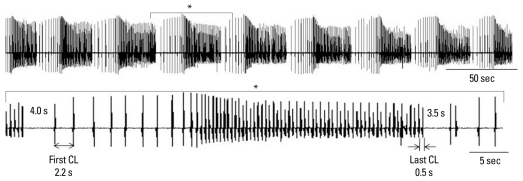
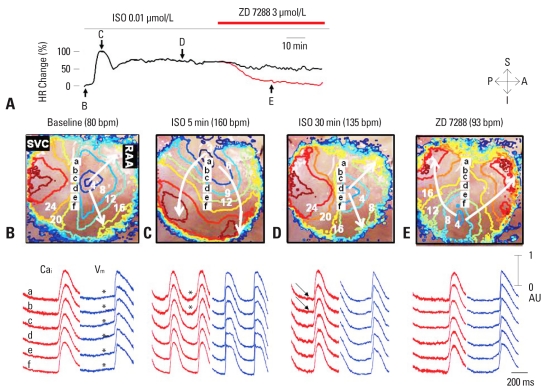
Recent evidence indicates that the voltage clock (cyclic activation and deactivation of membrane ion channels) and Ca(2+) clocks (rhythmic spontaneous sarcoplasmic reticulum Ca(2+) release) jointly regulate sinoatrial node (SAN) automaticity. However, the relative importance of the voltage clock and Ca(2+) clock for pacemaking was not revealed in sick sinus syndrome. Previously, we mapped the intracellular calcium (Ca(i)) and membrane potentials of the normal intact SAN simultaneously using optical mapping in Langendorff-perfused canine right atrium. We demonstrated that the sinus rate increased and the leading pacemaker shifted to the superior SAN with robust late diastolic Ca(i) elevation (LDCAE) during β-adrenergic stimulation. We also showed that the LDCAE was caused by spontaneous diastolic sarcoplasmic reticulum (SR) Ca(2+) release and was closely related to heart rate changes. In contrast, in pacing induced canine atrial fibrillation and SAN dysfunction models, Ca(2+) clock of SAN was unresponsiveness to β-adrenergic stimulation and caffeine. Ryanodine receptor 2 (RyR2) in SAN was down-regulated. Using the prolonged low dose isoproterenol together with funny current block, we produced a tachybradycardia model. In this model, chronically elevated sympathetic tone results in abnormal pacemaking hierarchy in the right atrium, including suppression of the superior SAN and enhanced pacemaking from ectopic sites. Finally, if the LDCAE was too small to trigger an action potential, then it induced only delayed afterdepolarization (DAD)-like diastolic depolarization (DD). The failure of DAD-like DD to consistently trigger a sinus beat is a novel mechanism of atrial arrhythmogenesis. We conclude that dysfunction of both the Ca(2+) clock and the voltage clock are important in sick sinus syndrome.
最近的证据表明,电压钟(膜离子通道的循环激活和失活)和 Ca(2+)钟(节律性自发性肌浆网 Ca(2+)释放)共同调节窦房结(SAN)的自律性。然而,在病态窦房结综合征中,电压钟和 Ca(2+)钟对起搏的相对重要性尚未揭示。之前,我们使用在 Langendorff 灌注犬右心房中进行的光学映射同时绘制了正常完整 SAN 的细胞内钙(Ca(i))和膜电位。我们证明,在β-肾上腺素刺激期间,窦性频率增加,并且随着强烈的舒张晚期 Ca(i)升高(LDCAE),主导起搏点移至上 SAN。我们还表明,LDCAE 是由自发性舒张期肌浆网(SR)Ca(2+)释放引起的,并且与心率变化密切相关。相比之下,在起搏诱导的犬心房颤动和 SAN 功能障碍模型中,SAN 的 Ca(2+)钟对β-肾上腺素刺激和咖啡因无反应。SAN 中的 Ryanodine 受体 2(RyR2)下调。使用延长的低剂量异丙肾上腺素和有趣的电流阻断,我们产生了心动过速-心动过缓模型。在该模型中,慢性升高的交感神经张力导致右心房中的异常起搏层次结构,包括抑制上 SAN 和增强异位起搏点的起搏。最后,如果 LDCAE 太小而不能引发动作电位,则只会引起延迟后除极(DAD)样舒张去极化(DD)。DAD 样 DD 不能持续引发窦性搏动是心房心律失常发生的一种新机制。我们得出结论,Ca(2+)钟和电压钟的功能障碍在病态窦房结综合征中都很重要。