Department of Biochemistry and Molecular Biology, Oregon Health and Science University, Portland, OR 97239, USA.
J Gen Physiol. 2011 Mar;137(3):299-314. doi: 10.1085/jgp.201010557. Epub 2011 Feb 14.
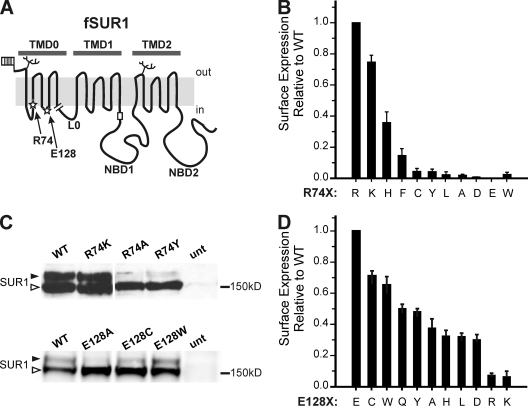
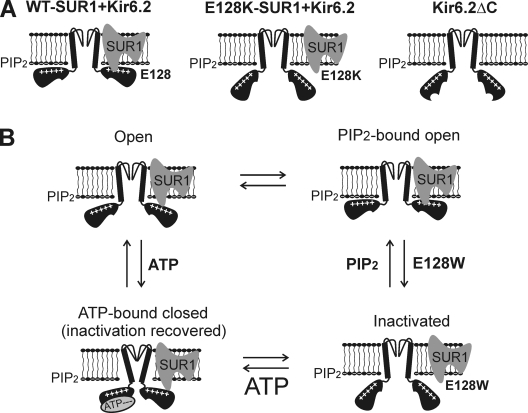
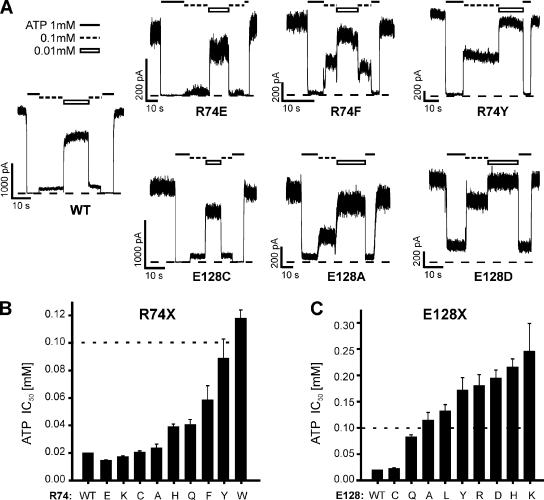
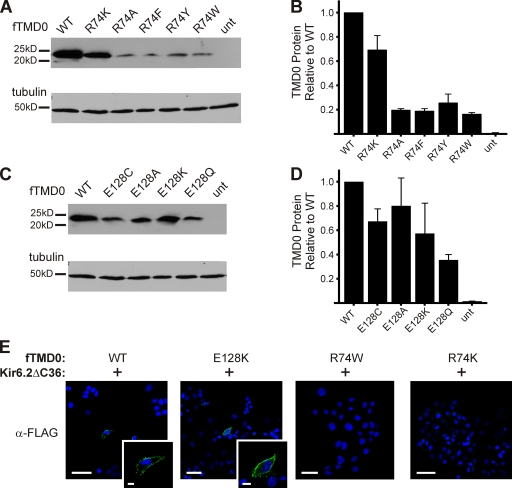
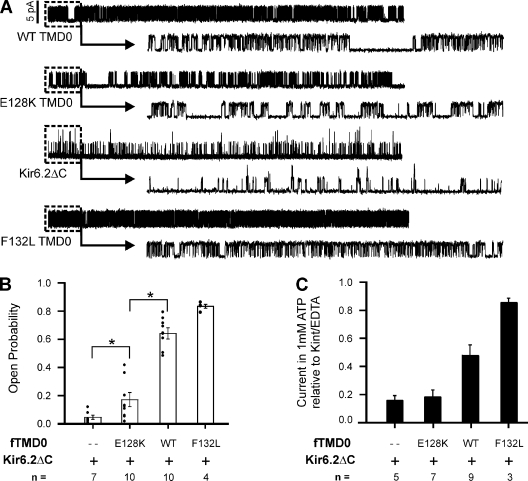
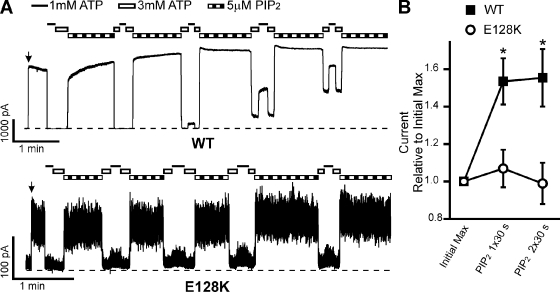
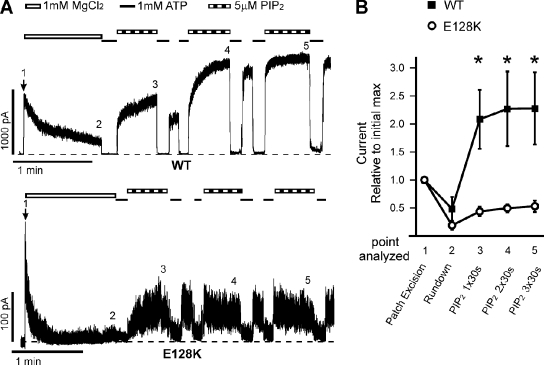
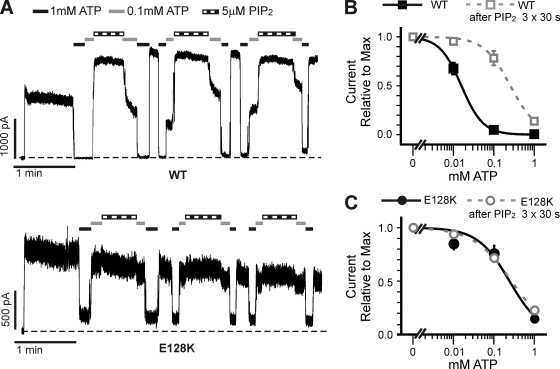
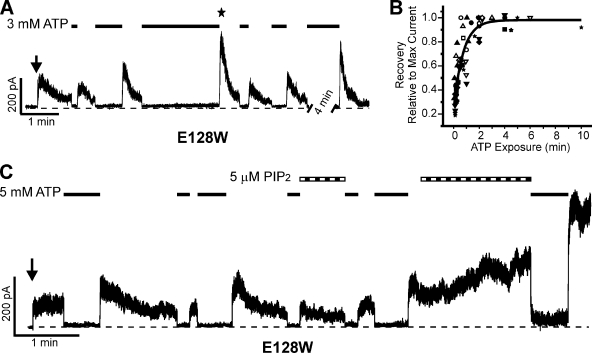
Functional integrity of pancreatic adenosine triphosphate (ATP)-sensitive potassium (K(ATP)) channels depends on the interactions between the pore-forming potassium channel subunit Kir6.2 and the regulatory subunit sulfonylurea receptor 1 (SUR1). Previous studies have shown that the N-terminal transmembrane domain of SUR1 (TMD0) interacts with Kir6.2 and is sufficient to confer high intrinsic open probability (P(o)) and bursting patterns of activity observed in full-length K(ATP) channels. However, the nature of TMD0-Kir6.2 interactions that underlie gating modulation is not well understood. Using two previously described disease-causing mutations in TMD0 (R74W and E128K), we performed amino acid substitutions to study the structural roles of these residues in K(ATP) channel function in the context of full-length SUR1 as well as TMD0. Our results revealed that although R74W and E128K in full-length SUR1 both decrease surface channel expression and reduce channel sensitivity to ATP inhibition, they arrive there via distinct mechanisms. Mutation of R74 uniformly reduced TMD0 protein levels, suggesting that R74 is necessary for stability of TMD0. In contrast, E128 mutations retained TMD0 protein levels but reduced functional coupling between TMD0 and Kir6.2 in mini-K(ATP) channels formed by TMD0 and Kir6.2. Importantly, E128K full-length channels, despite having a greatly reduced P(o), exhibit little response to phosphatidylinositol 4,5-bisphosphate (PIP(2)) stimulation. This is reminiscent of Kir6.2 channel behavior in the absence of SUR1 and suggests that TMD0 controls Kir6.2 gating by modulating Kir6.2 interactions with PIP(2). Further supporting this notion, the E128W mutation in full-length channels resulted in channel inactivation that was prevented or reversed by exogenous PIP(2). These results identify a critical determinant in TMD0 that controls Kir6.2 gating by controlling channel sensitivity to PIP(2). Moreover, they uncover a novel mechanism of K(ATP) channel inactivation involving aberrant functional coupling between SUR1 and Kir6.2.
胰腺三磷酸腺苷(ATP)敏感性钾(K(ATP))通道的功能完整性取决于孔形成钾通道亚基 Kir6.2 和调节亚基磺酰脲受体 1(SUR1)之间的相互作用。先前的研究表明,SUR1 的 N 端跨膜结构域(TMD0)与 Kir6.2 相互作用,足以赋予全长 K(ATP)通道中观察到的高固有开放概率(P(o))和爆发活动模式。然而,门控调节所依赖的 TMD0-Kir6.2 相互作用的性质尚不清楚。使用先前描述的 TMD0 中的两种致病突变(R74W 和 E128K),我们进行了氨基酸取代,以研究这些残基在全长 SUR1 以及 TMD0 背景下对 K(ATP)通道功能的结构作用。我们的结果表明,尽管全长 SUR1 中的 R74W 和 E128K 均降低了表面通道表达并降低了通道对 ATP 抑制的敏感性,但它们是通过不同的机制到达那里的。R74 的突变均匀降低了 TMD0 蛋白水平,表明 R74 对于 TMD0 的稳定性是必需的。相比之下,E128 突变保留了 TMD0 蛋白水平,但在由 TMD0 和 Kir6.2 形成的 mini-K(ATP)通道中,功能性 TMD0 和 Kir6.2 之间的偶联减少。重要的是,尽管 E128K 全长通道的 P(o)大大降低,但对磷脂酰肌醇 4,5-二磷酸(PIP(2))刺激几乎没有反应。这让人联想到缺乏 SUR1 时 Kir6.2 通道的行为,并表明 TMD0 通过调节 Kir6.2 与 PIP(2)的相互作用来控制 Kir6.2 门控。进一步支持这一观点,全长通道中的 E128W 突变导致通道失活,而外源性 PIP(2)可以阻止或逆转该失活。这些结果确定了 TMD0 中的一个关键决定因素,该决定因素通过控制通道对 PIP(2)的敏感性来控制 Kir6.2 门控。此外,它们揭示了一种涉及 SUR1 和 Kir6.2 之间异常功能偶联的新型 K(ATP)通道失活机制。