Department of Computer Science & Engineering, University of Connecticut, 371 Fairfield Rd, Unit 2155, Storrs, CT 06269-2155, USA.
BMC Bioinformatics. 2011 Feb 15;12 Suppl 1(Suppl 1):S53. doi: 10.1186/1471-2105-12-S1-S53.
Recent technology advances have enabled sequencing of individual genomes, promising to revolutionize biomedical research. However, deep sequencing remains more expensive than microarrays for performing whole-genome SNP genotyping.
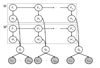
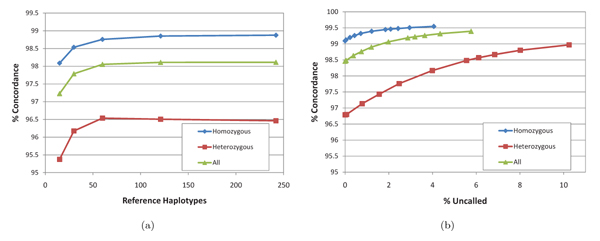
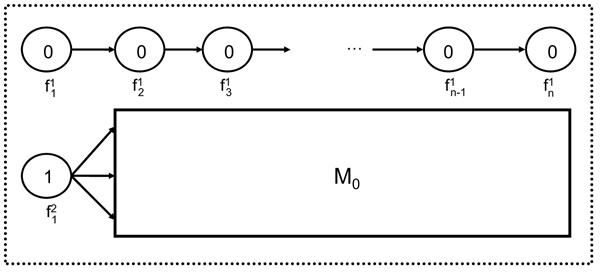
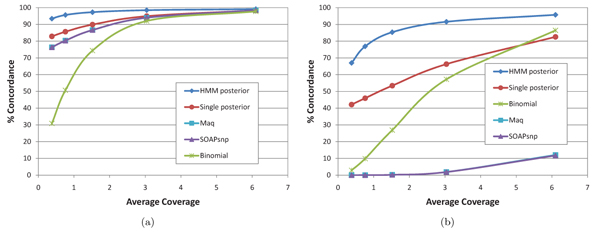
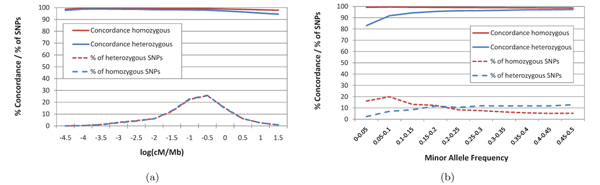
In this paper we introduce a new multi-locus statistical model and computationally efficient genotype calling algorithms that integrate shotgun sequencing data with linkage disequilibrium (LD) information extracted from reference population panels such as Hapmap or the 1000 genomes project. Experiments on publicly available 454, Illumina, and ABI SOLiD sequencing datasets suggest that integration of LD information results in genotype calling accuracy comparable to that of microarray platforms from sequencing data of low-coverage. A software package implementing our algorithm, released under the GNU General Public License, is available at http://dna.engr.uconn.edu/software/GeneSeq/.
Integration of LD information leads to significant improvements in genotype calling accuracy compared to prior LD-oblivious methods, rendering low-coverage sequencing as a viable alternative to microarrays for conducting large-scale genome-wide association studies.
最近的技术进步使得对个体基因组进行测序成为可能,有望彻底改变生物医学研究。然而,深度测序在进行全基因组 SNP 基因分型方面仍然比微阵列昂贵。
在本文中,我们介绍了一种新的多基因座统计模型和计算高效的基因型调用算法,该算法将测序数据与来自参考人群面板(如 Hapmap 或 1000 基因组计划)的连锁不平衡(LD)信息进行整合。对公开的 454、Illumina 和 ABI SOLiD 测序数据集进行的实验表明,整合 LD 信息可使基因型调用准确性与低覆盖测序的微阵列平台相当。我们的算法的软件包已在 GNU 通用公共许可证下发布,可在 http://dna.engr.uconn.edu/software/GeneSeq/ 上获得。
与先前的 LD 忽略方法相比,整合 LD 信息可显著提高基因型调用准确性,从而使低覆盖测序成为进行大规模全基因组关联研究的微阵列的可行替代方法。