Bovine Functional Genomics Laboratory, ANRI, USDA-ARS, Beltsville, Maryland 20705, USA.
BMC Genomics. 2011 Feb 23;12:127. doi: 10.1186/1471-2164-12-127.
Copy number variation (CNV) represents another important source of genetic variation complementary to single nucleotide polymorphism (SNP). High-density SNP array data have been routinely used to detect human CNVs, many of which have significant functional effects on gene expression and human diseases. In the dairy industry, a large quantity of SNP genotyping results are becoming available and can be used for CNV discovery to understand and accelerate genetic improvement for complex traits.
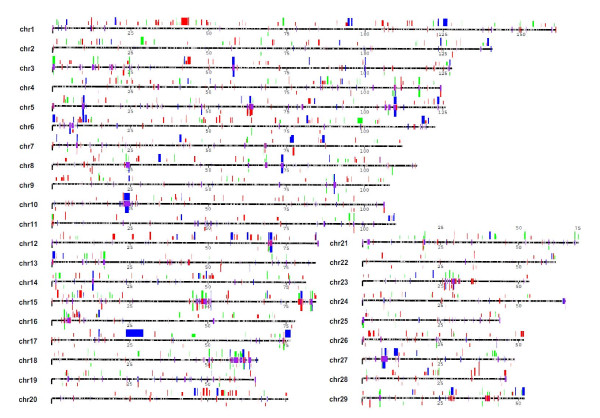
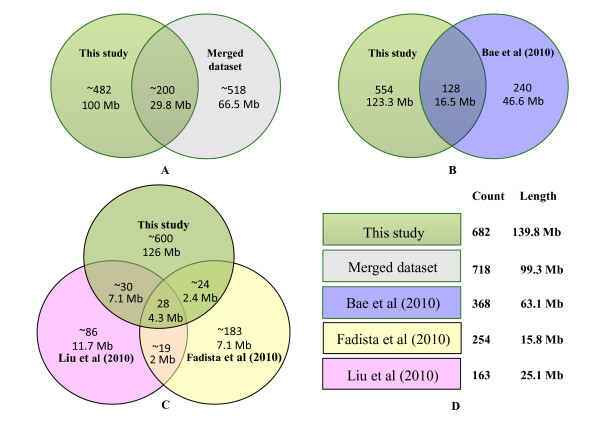
We performed a systematic analysis of CNV using the Bovine HapMap SNP genotyping data, including 539 animals of 21 modern cattle breeds and 6 outgroups. After correcting genomic waves and considering the pedigree information, we identified 682 candidate CNV regions, which represent 139.8 megabases (4.60%) of the genome. Selected CNVs were further experimentally validated and we found that copy number "gain" CNVs were predominantly clustered in tandem rather than existing as interspersed duplications. Many CNV regions (56%) overlap with cattle genes (1,263), which are significantly enriched for immunity, lactation, reproduction and rumination. The overlap of this new dataset and other published CNV studies was less than 40%; however, our discovery of large, high frequency (> 5% of animals surveyed) CNV regions showed 90% agreement with other studies. These results highlight the differences and commonalities between technical platforms.
We present a comprehensive genomic analysis of cattle CNVs derived from SNP data which will be a valuable genomic variation resource. Combined with SNP detection assays, gene-containing CNV regions may help identify genes undergoing artificial selection in domesticated animals.
拷贝数变异(CNV)是遗传变异的另一个重要来源,与单核苷酸多态性(SNP)互补。高密度 SNP 芯片数据已被常规用于检测人类 CNV,其中许多对基因表达和人类疾病具有显著的功能影响。在乳品行业,大量的 SNP 基因分型结果正在变得可用,并且可用于 CNV 发现,以了解和加速复杂性状的遗传改良。
我们使用 Bovine HapMap SNP 基因分型数据进行了 CNV 的系统分析,包括 21 个现代牛品种的 539 个动物和 6 个外群。在纠正基因组波动并考虑系谱信息后,我们鉴定了 682 个候选 CNV 区域,代表基因组的 139.8 兆碱基(4.60%)。选择的 CNV 进一步进行了实验验证,我们发现拷贝数“获得”CNV 主要是串联聚集的,而不是作为分散的重复存在。许多 CNV 区域(56%)与牛基因(1,263 个)重叠,这些基因在免疫、泌乳、繁殖和反刍方面显著富集。这个新数据集与其他已发表的 CNV 研究的重叠不到 40%;然而,我们发现的大的、高频(调查动物的>5%)CNV 区域与其他研究有 90%的一致性。这些结果突出了技术平台之间的差异和共同点。
我们提出了一个基于 SNP 数据的牛 CNV 的全面基因组分析,这将是一个有价值的基因组变异资源。与 SNP 检测分析相结合,含有基因的 CNV 区域可能有助于识别在驯化动物中经历人工选择的基因。