Department of Mathematics, Statistics and Computer Science, University of Illinois at Chicago, Chicago, Illinois, United States of America.
PLoS One. 2011 Mar 2;6(3):e17293. doi: 10.1371/journal.pone.0017293.
Most existing methods for phylogenetic analysis involve developing an evolutionary model and then using some type of computational algorithm to perform multiple sequence alignment. There are two problems with this approach: (1) different evolutionary models can lead to different results, and (2) the computation time required for multiple alignments makes it impossible to analyse the phylogeny of a whole genome. This motivates us to create a new approach to characterize genetic sequences.
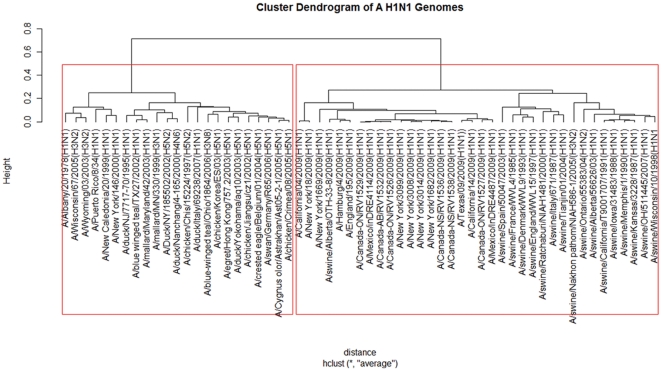
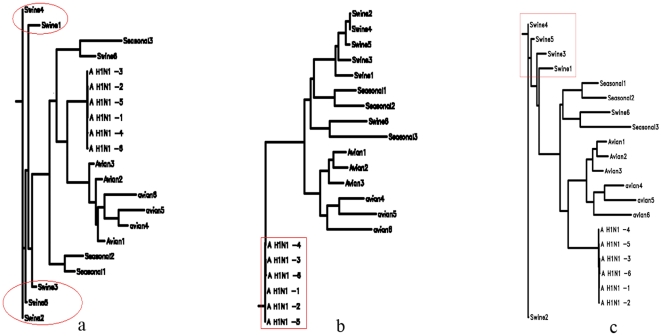
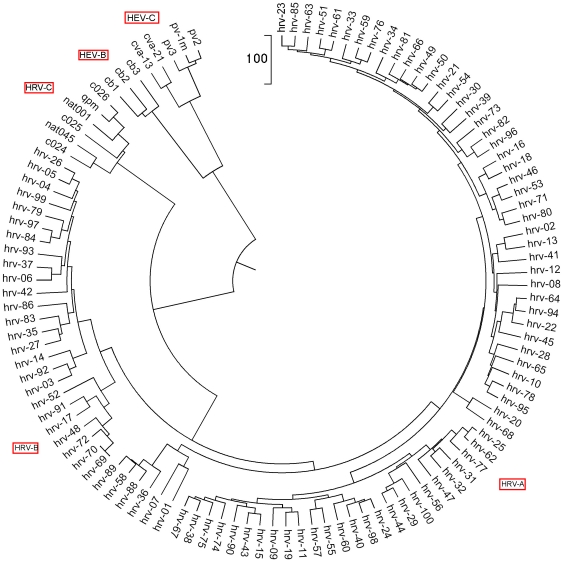
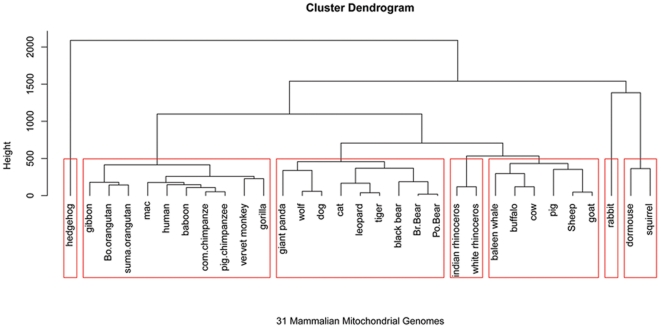
To each DNA sequence, we associate a natural vector based on the distributions of nucleotides. This produces a one-to-one correspondence between the DNA sequence and its natural vector. We define the distance between two DNA sequences to be the distance between their associated natural vectors. This creates a genome space with a biological distance which makes global comparison of genomes with same topology possible. We use our proposed method to analyze the genomes of the new influenza A (H1N1) virus, human rhinoviruses (HRV) and mammalian mitochondrial. The result shows that a triple-reassortant swine virus circulating in North America and the Eurasian swine virus belong to the lineage of the influenza A (H1N1) virus. For the HRV and mammalian mitochondrial genomes, the results coincide with biologists' analyses.
Our approach provides a powerful new tool for analyzing and annotating genomes and their phylogenetic relationships. Whole or partial genomes can be handled more easily and more quickly than using multiple alignment methods. Once a genome space has been constructed, it can be stored in a database. There is no need to reconstruct the genome space for subsequent applications, whereas in multiple alignment methods, realignment is needed to add new sequences. Furthermore, one can make a global comparison of all genomes simultaneously, which no other existing method can achieve.
大多数现有的系统发育分析方法都涉及开发进化模型,然后使用某种类型的计算算法来执行多重序列比对。这种方法有两个问题:(1)不同的进化模型可能会导致不同的结果,(2)多重比对所需的计算时间使得不可能分析整个基因组的系统发育。这促使我们创造一种新的方法来描述遗传序列。
我们为每个 DNA 序列关联一个基于核苷酸分布的自然向量。这在 DNA 序列与其自然向量之间产生了一一对应的关系。我们将两个 DNA 序列之间的距离定义为它们相关联的自然向量之间的距离。这创建了一个具有生物距离的基因组空间,使得具有相同拓扑结构的基因组的全局比较成为可能。我们使用我们提出的方法来分析新型甲型流感 (H1N1) 病毒、人类鼻病毒 (HRV) 和哺乳动物线粒体的基因组。结果表明,在北美循环的三重重配猪病毒和欧亚猪病毒属于甲型流感 (H1N1) 病毒谱系。对于 HRV 和哺乳动物线粒体基因组,结果与生物学家的分析一致。
我们的方法为分析和注释基因组及其系统发育关系提供了一种强大的新工具。与使用多重比对方法相比,可以更轻松、更快速地处理整个或部分基因组。一旦构建了基因组空间,就可以将其存储在数据库中。与多重比对方法不同,不需要重新构建基因组空间来添加新序列。此外,可以同时对所有基因组进行全局比较,这是其他现有方法无法实现的。