Oregon Clinical and Translational Research Institute, Oregon Health & Science University, Portland, Oregon, United States of America.
PLoS One. 2011 Mar 24;6(3):e17820. doi: 10.1371/journal.pone.0017820.
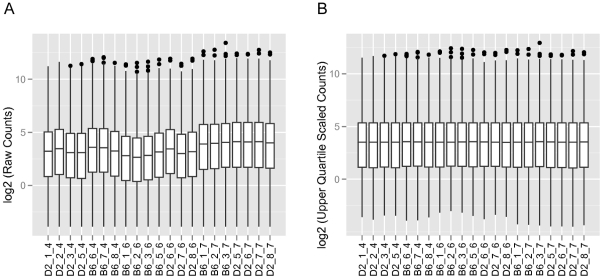
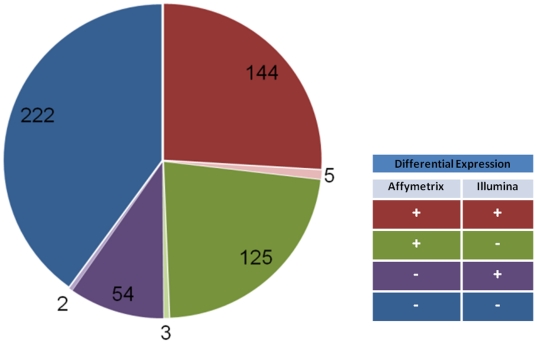
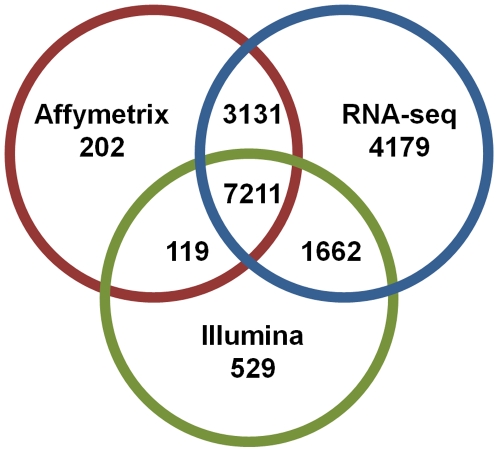
C57BL/6J (B6) and DBA/2J (D2) are two of the most commonly used inbred mouse strains in neuroscience research. However, the only currently available mouse genome is based entirely on the B6 strain sequence. Subsequently, oligonucleotide microarray probes are based solely on this B6 reference sequence, making their application for gene expression profiling comparisons across mouse strains dubious due to their allelic sequence differences, including single nucleotide polymorphisms (SNPs). The emergence of next-generation sequencing (NGS) and the RNA-Seq application provides a clear alternative to oligonucleotide arrays for detecting differential gene expression without the problems inherent to hybridization-based technologies. Using RNA-Seq, an average of 22 million short sequencing reads were generated per sample for 21 samples (10 B6 and 11 D2), and these reads were aligned to the mouse reference genome, allowing 16,183 Ensembl genes to be queried in striatum for both strains. To determine differential expression, 'digital mRNA counting' is applied based on reads that map to exons. The current study compares RNA-Seq (Illumina GA IIx) with two microarray platforms (Illumina MouseRef-8 v2.0 and Affymetrix MOE 430 2.0) to detect differential striatal gene expression between the B6 and D2 inbred mouse strains. We show that by using stringent data processing requirements differential expression as determined by RNA-Seq is concordant with both the Affymetrix and Illumina platforms in more instances than it is concordant with only a single platform, and that instances of discordance with respect to direction of fold change were rare. Finally, we show that additional information is gained from RNA-Seq compared to hybridization-based techniques as RNA-Seq detects more genes than either microarray platform. The majority of genes differentially expressed in RNA-Seq were only detected as present in RNA-Seq, which is important for studies with smaller effect sizes where the sensitivity of hybridization-based techniques could bias interpretation.
C57BL/6J(B6)和 DBA/2J(D2)是神经科学研究中最常用的两种近交系小鼠品系。然而,目前唯一可用的小鼠基因组完全基于 B6 品系序列。随后,寡核苷酸微阵列探针仅基于这一 B6 参考序列,由于其等位基因序列差异,包括单核苷酸多态性(SNP),使得它们在跨小鼠品系进行基因表达谱比较时的应用变得可疑。新一代测序(NGS)和 RNA-Seq 的应用为检测差异基因表达提供了一种明确的替代方案,而无需杂交技术固有的问题。使用 RNA-Seq,每个样本平均产生 2200 万个短测序读段,共 21 个样本(10 个 B6 和 11 个 D2),这些读段与小鼠参考基因组进行比对,允许在两种品系的纹状体中查询 16183 个 Ensembl 基因。为了确定差异表达,基于映射到外显子的读段应用“数字 mRNA 计数”。本研究比较了 RNA-Seq(Illumina GA IIx)与两种微阵列平台(Illumina MouseRef-8 v2.0 和 Affymetrix MOE 430 2.0),以检测 B6 和 D2 近交系小鼠之间纹状体差异基因表达。我们表明,通过使用严格的数据处理要求,RNA-Seq 确定的差异表达与 Affymetrix 和 Illumina 平台在更多情况下是一致的,而不是与单一平台一致的情况更多,并且在折叠变化方向上的不一致情况很少。最后,我们表明与基于杂交的技术相比,RNA-Seq 获得了更多的信息,因为 RNA-Seq 检测到的基因比任何一种微阵列平台都多。在 RNA-Seq 中差异表达的大多数基因仅在 RNA-Seq 中被检测到存在,这对于效应量较小的研究很重要,因为基于杂交的技术的敏感性可能会影响解释。